
Мониторинг проводимой терапии лимфопролиферативных

Лейкопения – это снижение количества лейкоцитов в единице объема крови.
Лейкопения может появиться как следствие различных заболеваний, в том числе и онкологических.
Лейкоциты – это клетки, имеющие важное значение для иммунной системы человека; белые кровяные тельца выполняют защитную функцию для организма от вирусов, бактерий и прочих агентов, способных причинить вред человеку и вызвать заболевания.
Норма содержания лейкоцитов в крови: от 4 до 9 х 109 клеток/ 1л. Если выявлено их снижение, то это может быть сигналом о подозрении на лейкопению.
- Острая
- Хроническая
- Легкая форма
- Средняя степень тяжести
- Тяжелая форма
Существуют различные виды лейкоцитов, обычно происходит снижение не всех, а только нескольких видов.
Причины возникновения лейкопении
- Метастазирование опухолевых клеток в костный мозг. Раковые клетки поражают ткани красного костного мозга, вызывая нарушения кроветворения и развитие тромбоцитопении, лейкопении, анемии.
- Аутоиммунная причина. Иммунитет человека атакует собственные белые кровяные тельца, ускоряя их разрушение. Такой процесс характерен для хронического лимфолейкоза.
- Прием цитостатических препаратов. Такие препараты оказывают разрушающее воздействие не только на делящиеся раковые клетки, но и на здоровые, особенно негативно цитостатики воздействуют на костный мозг.
- Лучевая терапия – этот метод лечения тоже негативно сказывается на процессе кроветворения в организме.
Опасность, которую несет лейкопения
Лейкоциты – это часть иммунной системы. Белые кровяные тельца атакуют чужеродные агенты, попадающие в организм, но при снижении лейкоцитов, иммунитет человека падает, защитные силы ослабевают. Организм становится более подвержен инфекционным заболеваниям.
Диагностика лимфопролиферативных заболеваний
Заболевание определяется с помощью общего анализа крови.Происходит подсчет содержания количества лейкоцитов в крови и соотношение различных видов лейкоцитов (лимфоциты, эозинофилы, базофилы, нейтрофилы, моноциты) в процентах.
Симптоматика при лейкопении может отсутствовать, особенно легкой степени заболевания. Первым симптомом может быть возникновение различных инфекций. У некоторых онкобольных наблюдается лихорадка, вызванная либо инфицированием, либо как реакция организма на прием химиопрепаратов, либо лихорадка имеет опухолевую причину.
- Головокружение
- Слабость, сонливость
- Бледность кожных покровов
- Кровоточивость слизистых тканей
- Появление синяков.
- Тяжелейшими осложнениями инфекционного характера являются сепсис и септический шок.
Лечение лимфопролиферативных заболеваний
Пациент, проходящий курс химиотерапии, должен находиться под постоянным наблюдением онколога, контролирующего уровень лейкоцитов в крови.
- Воздержаться от употребления сырой воды.
- Длительно варить мясо.
- Употреблять молоко и прочите жидкие продукты только из заводской упаковки, не на разлив.
- Обязательно тщательно мыть овощи и фрукты.
- Избегать контакта с больными людьми.
- Надевать медицинскую маску при нахождении в общественных местах. Заметив повышение температуры, незамедлительно обратиться к врачу.
Пациент, у которого наблюдается ярко выраженная симптоматика заболевания помещается в изолированную палату, а медицинский персонал, с которым больной имеет контакт, обязательно должен соблюдать все санитарные правила антисептики. Пациент может быть направлен в гематологическую клинику, специализирующуюся только на заболеваниях крови.
Больному вводят лекарственные препараты, стимулирующие образование лейкоцитов, дополнительно могут быть назначены витаминно-минеральные комплексы для поддержания организма.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Патогенез
- Симптомы
- Формы
- Диагностика
- Что нужно обследовать?
- Какие анализы необходимы?
- Дифференциальная диагностика
- Лечение
- Прогноз

Аутоиммунный лимфопролиферативный синдром (АЛПС) - заболевание, в основе развития которого лежат врожденные дефекты Fas-опосредоаанного апоптоза. Оно было описано в 1995 г., но еще с 60-х годов заболевание со схожим фенотипом было известно под названием синдром CanaLe-Smith.
Заболевание характеризуется хронической незлокачественной лимфопролиферацией и гипергаммаглобулинемией, которые могут сочетаться с различными аутоиммунными нарушениями.

[1], [2], [3], [4], [5]
Код по МКБ-10
Патогенез
Апоптоз, или физиологическая гибель клетки, является одним из неотъемлемых механизмов поддержания гомеостаза организма. Апоптоз развивается вследствие активации различных сигнальных механизмов. Особую роль в регуляции системы гемопоэза и иммунной системы играет апоптоз, опосредованный активацией Fas-рецепторов (CD95) при их взаимодействии с соответствующим лигандом (Fas-лиганд, FasL). Fas представлен на различных гемопоэтических клетках, высокая экспрессия Fas рецептора характерна для активированных лимфоцитов. Fasl-экспрессируется, главным образом, CD8+T-лимфоцитами.
Активация Fas рецептора влечет за собой ряд последовательных внутриклеточных процессов, итогом которых является дезорганизация ядра клетки, денатурация ДНК, изменения мембраны клетки, приводящие к ее распаду на ряд фрагментов без выброса во внеклеточную среду лизосомальных ферментов и без индукции воспаления. В передаче апоптотического сигнала к ядру участвует ряд ферментов, называемых каспазами, в том числе каспаза 8 и каспаза 10.
Fas-опосредованный апоптоз играет важную роль в элиминации клеток с соматическими мутациями, аутореамтивных лимфоцитов, а также лимфоцитов, выполнивших свою роль в процессе нормального иммунного ответа. Нарушение апоптоза Т-лимфоцитов приводит к экспансии активированных Т-клеток, а также так называемых двойных негативных Т-лимфоцитов, которые экспрессируют Т-клеточный рецептор с a/b цепями (TCRa/b), но не имеют ни CD4, ни CD8 молекул. Дефект программируемой гибели В-клеток в совокупности с повышением уровня интерлейкина 10 (IL-10) приводят к гипергаммаглобулинемии и повышению выживаемости аутореактивных В-лимфоцитов. Клиническими последствиями являются избыточное накопление лимфоцитов в крови и лимфоидных органах, увеличение риска аутоиммунных реакций и опухолевого роста.
К настоящему времени выявлено несколько молекулярных дефектов, приводящих к нарушению апоптоза и развитию АЛЛС. Это мутации в генах Fas, FasL, каспазы 8 и каспаэы 10.
[6], [7], [8], [9], [10], [11], [12], [13]
Симптомы аутоиммунного лимфопролиферативного синдрома
АЛПС отличается большой вариабельностью спектра клинических проявлений и тяжести течения, и возраст клинической манифестации также может колебаться в зависимости от выраженности симптоматики. Известны случаи дебюта аутоиммунных проявлений во взрослом возрасте, когда и был диагностирован АЛПС. Проявления лимфопролиферативного синдрома присутствуют с рождения в виде увеличения всех групп лимфоузлов (периферических, внутригрудных, внутрибрюшных), увеличения размеров селезенки, а часто и печени. Размеры лимфоидных органов могут изменяться в течение жизни, иногда отмечено их нарастание при интеркуррентных инфекциях. Лимфоузлы имеют обычную консистенцию, иногда плотноваты; безболезненны. Известны случаи резко выраженных проявлений гиперпластического синдрома, имитирующих лимфому, с увеличением периферических лимфоузлов, приводящим к деформации шеи, гиперплазией внутригрудных лимфоузлов вплоть до развития синдрома сдавления и дыхательной недостаточности. Описаны лимфоидные инфильтраты в легких. Однако во многих случаях проявления гиперпластического синдрома не столь драматичны, и они остаются незамеченными врачами и родителями. Степень выраженности спленомегалии также весьма вариабельна.
Тяжесть течения заболевания определяется главным образом аутоиммунными проявлениями, которые могут развиться в любом возрасте. Чаще всего встречаются различные иммунные гемопатии - нейтропения, тромбоцитопения, гемолитическая анемия, которые могут сочетаться в виде двух- и трехростковых цитопений. Может иметь место единичный эпизод иммунной цитопении, но зачастую они носят хронический или рецидивирующий характер.
Из других, более редких аутоиммунных проявлений, могут наблюдаться аутоиммунный гепатит, артрит, сиаладенит, воспалительные заболевания кишечника, узловатая эритема, панникулит, увеит, синдром Guiltain-Barre. Кроме того, могут наблюдаться различные кожные сыпи, преимущественно уртикарные, субфебрилитет или лихорадка без связи с инфекционным процессом.
У больных аутоиммунным лимфопролиферативным синдромом увеличена частота развития злокачественных опухолей по сравнению с популяцией. Описаны случаи гемобластозов, лимфом и солидных опухолей (карцинома печени, желудка).
[14]
Формы
В 1999 г. предложена рабочая классификация аутоиммунного лимфопролиферативного синдрома, основанная на типе дефекта апоптоза:
- ALP5 0 - полный дефицит CD95, являющийся следствием гомозиготной нуль-мутации (homozygous nuLl mutation) в гене Fas/CD95;
- ALPS I - дефект передачи сигнала через Fas-рецептор.
- При этом ALPS la является следствием дефекта Fas-рецептора (гетерозиготная мутация в гене Fas);
- ALPS lb является следствием дефекта Fas-лиганда (FasL), связанного с мутацией в соответствующем гене - FASLG/CD178;
- ALPS Ic является следствием только что выявленной гомозиготной мутации в гене FA5LG/CD178;
- ALPS II - дефект внутриклеточной передачи сигнала (мутация в гене каспазы 10 - ALPS IIа, в гене каспазы 8 - ALPS IIb);
- ALPS III - молекулярный дефект не установлен.
АЛПС 0 типа - полный дефицит CD95 - описан всего лишь у нескольких пациентов, Поскольку гетерозиготные члены семей не имеют фенотипа АЛПС, была предложена гипотеза о аутосомно-рецессивном типе наследования. Однако, неопубликованные данные о наблюдении за семьей, в которой был выявлен пациент с АЛПС 0, не полностью согласуются с данным утверждением. Ученые выяснили, что многие, если не все, мутации являются доминантными, и что если они оказываются гомозиготными, это приводит к более выраженному фенотипу заболевания.
При АЛПС I типа тип наследования - аутосомно-доминантный, с неполной пенетрантностью и вариабельной экспрессивностью. В частности, при АЛПС1а описаны случаи гомозиготности или сочетанной гетерозиготности, при которой определяются различные мутации гена Fas в обеих аллелях. Эти случаи характеризовались тяжелым течением с пренатальной или неонатальной манифестацией (водянка плода, гепатоспленомегалия, анемия, тромбоцитопения). Кроме того, выявлена корреляция выраженности клинической симптоматики с типом мутации в гене Fas; для мутации во внутриклеточном домене характерно более тяжелое течение. Всего в мире описано более 70 пациентов с ALPS la. Мутация FasL была впервые описана у пациента с клиническими проявлениями системной красной волчанки и хронической лимфопролиферацией. Она была категоризирована как ALPS lb, хотя фенотип не полностью отвечал критериям классического аутоиммунного лимфопролиферативного синдрома (двойные негативные Т-клетки и спленомегалия отсутствовали). Первая гомозиготная мутация А247Е в гене FasL (внеклеточный домен) была описана недавно, в 2006 году, Del-Rey M et al. у пациента с нелетальным АЛПС, что говорит о важной роли терминального домена FasL C0OH во взаимодействии Fas/FasL. Авторы предлагают в действующую классификацию аутоиммунного лимфопролиферативного синдрома внести подгруппу ALPS Ic.
АЛПС II типа наследуется по аутосомно-рецессивному типу, и у многих пациентов с этим типом заболевания отмечалась типичная клиническая и иммунологическая АЛПС, включая нарушенный Fas-опосредованный апоптоз, в реализации которого задействованы как каспаза 8 (вовлечена на ранних этапах межклеточной передачи сигнала на уровне взаимодействий TCR и BCR), так и каспаза 10 (вовлечена в апоптотический каскад на уровне всех известных рецепторов, которые индуцируют апоптоз лимфоцитов).
Более чем у 30 пациентов была выявлена клиническая картина АЛПС средней степени выраженности, включавшая в себя гипергаммаглобулинемию и повышенный уровень двойных негативных Т-клеток в крови, причем активированные лимфоциты пациентов с АЛПС III типа (именно так был назван этот синдром) показывали нормальную активацию Fas-опосредованного пути in vitro, и никаких молекулярных дефектов найдено не было. Возможно, причиной заболевания служат нарушений других апоптотических путей, опосредованных, например, Trail-R, DR3 или DR6. Интересным кажется наблюдение R. Qementi о выявлении мутации N252S в гене перфорина (PRF1) у больного с АЛПС III типа, у которого наблюдалось существенное снижение НК-активности. При этом автор отмечает, что значительная разница между частотой обнаружения N252S у пациентов с АЛПС (2 из 25) и частотой ее выявления в группе контроля (1 из 330) заставляет предполагать ее связь с развитием АЛПС в итальянской популяции. С другой стороны, F. Rieux-Laucat отмечает, что данный вариант мутации PRF1 выявлялся им у 18% здоровых и у 10% больных АЛПС (неопубликованные данные). И, кроме того, наряду с полиморфизмом N252S, им найдена мутация гена Fas у пациента с АЛПС и его здорового отца, что, по мнению F.Rieux-Laucat, говорит о непатогенности гетерозиготной мутации N252S в гене перфорина, описанной несколько ранее R. Qementi у пациента с АЛПС (мутация Fas) и крупноклеточной В-лимфомой. Таким образом, вопрос о причинах возникновения АЛПС III типа на сегодняшний день остается открытым.
[15], [16], [17], [18]
В теле человека имеются не только кровеносные, но и так называемые "белые" сосуды. Известны они были довольно давно, а в середине 18 столетия знания о лимфатической системе стали более обширными. К сожалению, нередко встречаются лимфопролиферативные заболевания, а возникнуть они могут в любом органе.
Лимфатическая система
Она выполняет в функционировании человека довольно важную роль: благодаря лимфатической системе происходит транспортировка полезных веществ, удаляется лишняя межтканевая жидкость. Еще одна немаловажная способность – это обеспечение иммунитета. Жидкость, которая выполняет данные задания, называется лимфой. Она имеет прозрачный цвет, в составе преобладают лимфоциты. Самой небольшой структурной единицей системы являются капилляры. Они переходят в сосуды, которые бывают как внутриорганными, так и внеорганными. Их строение включает и клапаны, что предотвращают обратный ток жидкости. Самые большие лимфатические сосуды имеют название коллекторы. Именно в них накапливается жидкость от внутренних органов и других больших частей тела. Еще одна составляющая, которую имеет лимфатическая система (фото расположено внизу), – узлы. Это круглые образования, которые имеют разный диаметр (от полумиллиметра до 5 сантиметров). Расположены они группами на пути сосудов. Основная функция – фильтрация лимфы. Именно здесь она очищается от вредных микроорганизмов.
Лимфатические органы
Частью лимфатической системы человека являются также и следующие органы: миндалины, вилочковая железа (тимус), селезенка, костный мозг. Лимфоциты, которые формируются в тимусе, имеют название Т-клетки. Их особенностью является непрерывная циркуляция между лимфой и кровью. Частицы, которые образовываются в костном мозге, называются В-клетками. Оба типа после созревания разносятся по организму. В-клетки остаются в лимфоидных органах. На этом их миграция прекращается. В брюшной полости размещается еще один крупный орган, который является неотъемлемой частью лимфатической системы, – это селезенка. Состоит она из двух частей, одна из них (белая пульпа) генерирует антитела.
Лимфопролиферативное заболевание. Что это такое
Возможные причины возникновения
Среди причин, способных вызвать лимфоприлиферативные заболевания, выделяют определенную группу вирусов. Также не последнюю роль играет и фактор наследственности. Заболевания кожи, которые длятся значительное время (например, псориаз) могут спровоцировать рост злокачественных новообразований. Ну и, конечно, существенно влияет на данный процесс излучение. Радиация, некоторые аллергены, токсические вещества активизируют процесс разрастания клеток.
Лимфомы. Диагностика
Одна из разновидностей злокачественных новообразований лимфатической системы – это лимфома. Симптомы на начальных стадиях могут быть не сильно выражены.
Наблюдается увеличение лимфатических узлов, которые не являются болезненными. Еще один яркий признак – усталость, причем в довольно большой степени. Пациент может жаловаться на повышенную потливость в ночное время, значительную и резкую потерю массы тела. Возможен также и зуд, красные пятна. Температура тела иногда повышается, особенно по вечерам. Насторожить такие симптомы должны тогда, если они не исчезают спустя несколько недель. Для эффективного лечения очень важно определить тип лимфомы. При диагностике учитывают место расположения, внешний вид опухоли, вид белка, что находится на ее поверхности. Специалист назначает полное медицинское исследование, анализ крови на раковые клетки, проводится диагностика внутренних органов. Для большей информативности необходима биопсия. Под микроскопом пораженные клетки имеют специфический вид.
Лечение лимфомы
Методы лечения данного заболевания следующие. Для уничтожения новообразования используют химиотерапию или радиотерапию (с помощью рентгеновских лучей). Используется комбинация препаратов, они распространяются в организме и могут уничтожить также и те клетки, которые не удалось диагностировать. После проведения химиотерапии поражается и костный мозг, поэтому может понадобиться его пересадка. Осуществляют ее как из материала донора, так и непосредственно из собственного костного мозга пациента (предварительно его извлекают до начала процедур). Лимфопролиферативные заболевания поддаются и биологической терапии, но она носит преимущественно экспериментальный характер. Базируется на применении веществ, что синтезируются из клеток пациента. Для достижения хорошего результата необходимо тщательно следовать указаниям лечащего врача, вовремя принимать препараты, уделить должное внимание питанию.

Лейкоз. Клиническая картина
Заболевание характеризуется изменением кроветворных клеток, при котором происходит замещение здоровых элементов костного мозга на пораженные. В крови значительно повышается уровень лимфоцитов. В зависимости от того, какие клетки были перерождены, выделяют болезнь лимфолейкоз (изменения лимфоцитов), миелолейкоз (поражены миелоциты). Определить вид болезни можно под микроскопом и по анализу белка. Лимфопролиферативное заболевание (что это такое, было описано выше) имеет в данном случае две формы протекания: хроническую и острую. Последняя проходит довольно тяжело. В этом случае необходимо незамедлительное лечение, так как клетки незрелые и не способны выполнять свои функции. Хроническая форма может длиться немало лет.
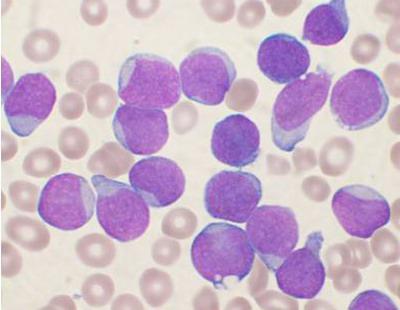
Хронические лимфопролиферативные заболевания
У людей старшего возраста нередко диагностируют хронический лимфолейкоз. Болезнь протекает довольно медленно, и только на поздних стадиях наблюдаются нарушения в процессе образования крови. К симптомам можно отнести увеличение лимфоузлов и селезенки, частые инфекционные заболевания, потерю веса, потливость. Нередко такие лимфопролиферативные заболевания обнаруживают случайно.
а) Терминология:
1. Сокращения:
• Посттрансплантационная лимфопролиферативная болезнь (ПТЛБ)
2. Синонимы:
• Посттрансплантационное лимфопролиферативное расстройство
3. Определение:
• Посттрансплантационное нарушение пролиферации В-клеток, Т-клеток или естественных клеток-киллеров, обычно связанное с инфицированием вирусом Эпштейна-Барр (ВЭБ)
• Также к данному заболеванию относятся опухоли, подобные лимфоме Ходжкина, если они возникают после трансплантации
б) Лучевые признаки посттрансплантационной лимфопролиферативной болезни (ПТЛБ):
1. Основные особенности:
• Оптимальный диагностический ориентир:
о Единичные или множественные узелки или объемные образования в легких
о ± лимфаденопатия средостения и корней легких; ± участки консолидации легочной ткани
• Локализация:
о Локализация зависит от того, какой орган был трансплантирован (например, легкое, печень, кишка, почка)
- Поражение органов грудной клетки чаще всего возникает после трансплантации легкого > сочетанной трансплантации сердца и легкого > сердца > печени > почки
![]()
(а) Пациент с посттрансплантационной лимфопролиферативной болезнью (ПТЛБ) после трансплантации печени. При рентгенографии органов грудной клетки в ПП проекции определяются узелки в обоих легких и крупное объемное образование в нижней доле правого легкого
(б) У этого же пациента при нативной КТ визуализируются множественные узелки в обоих легких и крупное объемное образование нижней доли правого легкого. Следует отметить наличие небольшого правостороннего плеврального выпота. Наиболее частым проявлением посттрансплантационной лимфопролиферативной болезни (ПТЛБ) при исследовании органов грудной клетки является наличие в легких единичных или множественных узелков и объемных образований.
4. Методы медицинской радиологии:
• ПЭТ:
о Облегчает выявление скрытого поражения экстралимфатических органов
о Более агрессивные формы ПТЛБ часто характеризуются более высоким уровнем поглощения ФДГ
5. Рекомендации к проведению лучевых исследований:
• Оптимальный метод лучевой диагностики:
о КТ используется для оценки как патологических изменений в легких, так и протяженности лимфаденопатии
• Рекомендации по выбору протокола:
о КТ с контрастным усилением шеи, органов грудной клетки, брюшной полости и малого таза: выявление распространенной формы заболевания, поражающей множество лимфатических узлов и экстралимфатических органов
о ПЭТ /КТ позволяет выявлять наличие скрытого заболевания
![]()
(а) При нативной КТ в обоих i легких определяются участки перибронхиальной консолидации легочной ткани, узловые уплотнения и небольшой плевральный выпот. Консолидация легочной ткани является нечастым проявлением ПТЛБ (10% от всех случаев).
(б) У пациента с посттрансплантационной лимфопролиферативной болезнью (ПТЛБ) при нативной КТ визуализируются объ емное образование нижней доли левого легкого и прилежащий к нему небольшой плевральный выпот. Наличие единичных узелков или объемных образований являются одним из наиболее частых проявлений посттрансплантационной лимфопролиферативной болезни (ПТЛБ) при исследовании органов грудной клетки.
в) Дифференциальная диагностика посттрансплантационной лимфопролиферативной болезни (ПТЛБ):
1. Пневмония грибковой этиологии:
• Симптомы инфекции
• Часто сопровождается плевральным выпотом
• По сравнению с ПТЛБ узелки имеют менее четкий контур и чаще содержат полости
• На ранних стадиях ангиоинвазивной аспергиллезной инфекции при КТ выявляется симптом ореола
2. Криптогенная организующаяся пневмония:
• У пациентов после трансплантации солидных органов часто поражаются периферические, перибронхиальные и базальные отделы легких, а после трансплантации стволовых клеток-верхние доли легких
• Участки консолидации легочной ткани округлой формы
• Часто выявляется симптом воздушной бронхограммы
• Высокая эффективность терапии стероидными препаратами
3. Рак легких:
• При КТ с контрастным усилением выявляется мягкотканное объемное образование, гетерогенно накапливающее контрастное вещество
• Может быть неотличим от посттрансплантационной лимфопролиферативной болезни
• Для постановки диагноза часто требуется выполнить биопсию
4. Метастазы:
• Множественные узелки и объемные образования в обоих легких
• Лимфаденопатия средостения и корней легких
• Считается, что у пациентов после трансплантации повышен риск развития злокачественных новообразований вследствие долговременной иммуносупрессивной терапии
5. Диффузное альвеолярное кровотечение:
• Обширное затемнение паренхимы легкого, обычно не узлового характера
• Может сопровождаться гемофтизом и анемией
в) Патоморфология посттрансплантационной лимфопролиферативной болезни (ПТЛБ):
1. Основные особенности:
• Этиология:
о Обычно связана с инфицированием вирусом Эпштейн-Барр (ВЭБ):
- Иммуносупрессивная терапия циклоспорином приводит к бесконтрольной пролиферации клеток, инфицированных ВЭБ
- Возможна моноклональная пролиферация и приобретение клетками злокачественного потенциала
- Почти 100% взрослого населения серопозитивны
- Вызывает инфекционный мононуклеоз у подростков и взрослых
- Основным фактором риска развития посттрансплантационной лимфопролиферативной болезни является серопозитивная реакция на ВЭБ
- Риск развития посттрансплантационной лимфопролиферативной болезни у серонегативного по ВЭБ реципиента, чей донор был серопозитивен, составляет 25-50%
• Считается, что посттрансплантационная лимфопролиферативная болезнь является промежуточной стадией между доброкачественной поликлональной гиперплазией лимфоидной ткани и лимфомой:
о Важно диагностировать данное заболевание до того, как оно трансформируется в агрессивную форму
• Экстралимфатическая посттрансплантационная лимфопролиферативная болезнь (2/3 пациентов):
о Голова и шея: кольцо Пирогова-Вальдейера (носоглотка, ротоглотка, миндалины)
о Утолщение стенок пищевода или кишки
о Спленомегалия
о Печень: изолированные объемные образования пониженной плотности (1 -4 см в диаметре) или диффузная инфильтрация
о Центральная нервная система: изолированные внутричерепные объемные образования
• Узловая посттрансплантационная лимфопролиферативная болезнь (1/3 пациентов):
о Лимфаденопатия: ретроперитонеальная, мезентериальная или подмышечная
2. Стадирование, определение степени дифференцировки и классификация опухолей:
• Классификация посттрансплантационной лимфопролиферативной болезни:
о Ранняя (реактивная гиперплазия)
о Полиморфная (поликлональная)
о Мономорфная (моноклональные В-кпетки или Т-клетки)
о Лимфома
3. Микроскопические особенности:
• Категории:
о Плазмоцитарная гиперплазия:
- Чаще всего встречается в ротоглотке или лимфатических узлах
- Обычно поликлональная
о Полиморфная В-клеточная гиперплазия и лимфома:
- Лимфатические узлы или экстралимфатические органы
- Обычно моноклональная
о Иммунобластная лимфома или множественная миелома:
- Распространенная форма заболевания
г) Клинические аспекты посттрансплантационной лимфопролиферативной болезни (ПТЛБ):
1. Проявления:
• Наиболее частые признаки:
о Симптомы зависят от органов, вовлеченных в опухолевый процесс
о Мононуклеозоподобный синдром (20%)
о Тонзиллит, синусит, средний отит
о Лимфаденопатия
• Другие симптомы:
о Симптомы отсутствуют: обнаруживают случайно при динамическом наблюдении
2. Демографические данные:
• Эпидемиология:
о Инцидентность зависит от типа трансплантации:
- Трансплантация комплекса внутренних органов (13-33%)
- Трансплантация кишки (7-11%)
- Трансплантация комплекса сердце-легкие (9,4%)
- Трансплантация легкого (2-8%)
- Трансплантация сердца, поджелудочной железы или печени (1-5%)
- Трансплантация стволовых клеток крови (В-клетки донора) или почек (
- Вернуться в оглавление раздела "Лучевая медицина"
Редактор: Искандер Милевски. Дата публикации: 5.2.2019
Читайте также: