
Раковые опухоли возникают в результате соматических мутаций в клетках тела
Мутационная теория канцерогенеза – это основная концепция о том, что онкологические заболевания являются генетическими, и в их основе лежат изменения в геноме.
В подавляющем большинстве рак развивается из всего лишь одной опухолевой клетки. Исходя из данной теории, к раку проводят накопленные мутации всего в одной клетке в специфических участках ДНК, которые приводят к развитию дефектных белков.
Развитие мутационной теории
Основоположником мутационной теории рака можно считать Теодора Бовери, высказавшего предположение о том, что нарушения в хромосомах приводят к образованию раковых клеток, сделано это было в 1914 году. После было обнаружено влияние ионизирующего излучения на мутации, а также множество других связей. На сегодняшний день учеными распознано более 100 генов, ассоциированных с раком.
По мнению специалистов, прямым доказательством мутагенной теории концерогенеза является наличие генов-супрессоров и протоонкогенов, изменение их структуры происходит в результате разнообразных мутационных событий, среди которых и точечные мутации. В результате развиваются злокачественные трансформации.
Гены-супрессоры — это гены, которые кодирующие белки, которые подавляют разрастание ткани в результате размножения клеток.
Протоонкогены — это гены, которые также кодируют белки. В случае нарушения их работы возможно развитие пухолевого процесса. Мутации, которые затронули такие клетки, повышают риск возникновения злокачественного новообразования.
Таким образом, гены-супрессоры опухолей, или антионкогены, — это гены, которые подавляют развитие опухоли.
Открытие и изучение клеточных протоонкогенов было впервые осуществлено при помощи специальных ретровирусов — восокоонкогенных РНК-вирусов, которые несут в своем составе трансформирующие гены. Молекулярными и биологическими методами было выявлено, что ДНК здоровых клеток некоторых видов аукариот содержит особые последовательности, которые схожи с вирусными онкогенами. Они получили название протоонкогены. Превращение таких последовательностей в онкогены обычно происходит как раз в результате мутаций.
В конце прошлого века были обнаружены гены, которые препятствуют делению клеток и выходу их из дифференцированного состояния. Из-за своей противоположной функции по отношению к онкогенам они были названы генами-супрессорами или антионкогенами злокачественности. Таким образом, такие гены совместно с протонкогенами создают сложную системуконтроля клеточной дифференцировки и пролиферации, а злокачественная трансформация создается при нарушении такой системы.
Гипотеза Кнудсона
В 80-х годах прошлого столетия ученый Альфред Кнудсон предложил теорию, которая сейчас известна как теория двойной мутации или двойного удара. Она объясняет механизм появления злокачественной опухоли сетчатки глаза. Данные статистического анализа доказали проявление различных форм опухоли глаза в результате двух событий: наследственной и ненаследственной мутации. Дальнейшие исследования подтвердили данную теорию, и на сегодняшний день она считается классической.
Современные представления в онкологии предполагают, что для завершения начавшегося процесса образования опухоли необходимо от трех до шести генетических повреждений. Данные молекулярно-генетических, экспериментальных (на основе трансгенных животных и трансформированных животных), клинических и эпидемиологических исследований полностью согласуются с данными представлениями.
Мутаторный фенотип
Возникновение онкологических заболеваний у людей значительно превышает теоретическое ожидание, если учесть случайное и независимое возникновение в опухолевой клетке мутаций. Чтобы объяснить данное противоречие была предложена модель мутаторного фенотипа. У человека известны 6 генов стабильности. Такие клетки с дефектом характеризуются увеличением частоты спонтанных мутаций от двукратного до шестидесятикратного повышения мутабельности.
Кроме мутационной теории мутагенеза, принимаемой за основу на сегодняшний день, существует и множество ее альтернативных вариантов.

Об этом мы поговорили с онкогинекологом, хирургом Владимиром Носовым, руководителем Клиники гинекологии и онкогинекологии Eвропейского медицинского центра – первой клиники в России, где персонализированная терапия онкогинекологических заболеваний стала стандартной практикой.
В своём нормальном состоянии эти гены участвуют в восстановлении ДНК после различных повреждений, тем самым защищая клетки от опухолевого перерождения. Если возникает мутация в этих генах, здоровые клетки оказываются не защищенными и сами могут становиться злокачественными. Вероятность заболеть раком груди при носительстве мутации гена BRCA 1/2 колоссальная — до 80%(в общей популяции у женщин без мутации — около 10-12%), риск заболеть раком яичников — до 40-45 %( в популяции —около 1,5%) .
В большинстве случаев назначение этих препаратов после первой линии химиотерапии обеспечивает ремиссию около 3 лет – это огромное достижение, еще никогда в онкогинекологии ремиссия при 3-4 стадии заболевания не продлевалась каким-либо лекарством на столь длительный срок.
Дальнейшие исследования позволили выяснить, что мутации могут быть не только герминогенными, то есть присутствующими во всех клетках организма. Дополнительные 15-20% мутаций генов BRCA происходят только в клетках опухоли, но в крови и других клетках организма их нет. Эти мутации называют соматическими. Они не передаются по наследству, не увеличивают риск развития других онкологических заболеваний, но пациенты, у которых обнаружены мутации в клетках опухоли, также являются кандидатами для лечения ингибиторами PARP.
В Институте онкологии EMC мы предлагаем всем пациентам с раком яичников провести полное секвенирование генов BRCA опухоли и крови. Это позволяет подобрать наиболее эффективную персонализированную терапию. Если речь идет о наследственной мутации – мы рекомендуем в обязательном порядке генетическое обследование детям, сестрам, братьям, родителям, а самим пациенткам-носителям мутации – также пройти дополнительный скрининг на рак молочных желез, риски которого колоссально повышены.
Плохое наследство
Наследственная мутация передается детям с вероятностью 50%, причем как по женской, так и по мужской линии. Носителям мы рекомендуем специальную программу наблюдения и профилактические мероприятия для снижения риска онкологических заболеваний, а также обсуждаем с ними вопросы сохранения репродуктивной функции.
Например, на днях я оперировал пациентку 57 лет с раком яичника. На плановой гистологии был подтвержден злокачественный характер опухоли. Мы провели генетическое исследование опухоли, выявили мутацию BRCA1. Затем было выполнено полное генетическое исследование по крови, чтобы понять, является ли мутация соматической (присутствующей только в опухоли) или герминогенной (наследственной). Выяснилось, что мутация наследственная. Мы рекомендовали пройти обследование двум дочерям пациентки, которые, к сожалению, унаследовали эту мутацию. Женщины-близнецы, им сейчас 31 год, обе еще не планировали беременность и роды. Я рекомендовал им обратиться к репродуктологу, провести стимуляцию и заморозить яйцеклетки, а в 35 лет, именно с этого возраста риски рака яичников начинают расти, удалить профилактически яичники и маточные трубы. В этом случае мы сохраняем матку, и в будущем они смогут выносить своих биологических детей.
Более того, во время ЭКО можно провести предимплантационную диагностику и подсадить эмбрионы, не унаследовавшие мутацию. Таким образом, будущее поколение уже будет защищено.
Рак эндометрия (рак тела матки) – самое распространенное онкогинекологическое заболевание у женщин. Сегодня подходы к его лечению также меняются благодаря персонализированной терапии.
До недавних пор считалось, что существует два типа рака эндометрия. Наиболее частый, первого типа, обычно возникает у полных пациентов, часто с сопутствующими диабетом и гипертонией. Второй – серозный, более агрессивный, не связанный с избытком эстрогенов. На основании клинической картины врачи принимали решение о необходимости дополнительного лечения после операции. Сегодня, благодаря лучшему пониманию биологии опухоли, мы знаем, что этих типов не два, а четыре. И для каждого из них предусмотрено определенное лечение. Чтобы определить, с каким типом рака эндометрия мы имеем дело, достаточно для начала провести иммуногистохимическое исследование.
Каждую опухоль эндометрия вне зависимости от стадии, мы тестируем на наличие определенных молекул, указывающих на благоприятный или менее благоприятный прогноз заболевания. Например, наличие мутации гена P53 говорит о менее благоприятном прогнозе. В этом случае мы рекомендуем не только наблюдение, но и дополнительное лечение с помощью химио-или лучевой терапии.
Некоторые раки матки, так же, как и некоторые раки яичников и молочной железы, имеют в своей основе генетический синдром – синдром Линча. Если мы находим проявления синдрома Линча в опухоли, мы направляем пациентов на полноценное генетическое тестирование. Это важно, потому что рак матки – не единственное заболевание, к которому предрасположены носители мутаций, вызывающих синдром Линча. В частности, у них повышен риск рака толстой кишки в молодом возрасте.
Часто первым возникает рак матки, через какое-то время развивается рак толстой кишки.
Поэтому носителям синдрома Линча рекомендуют начинать скрининг на рак кишки не в 45-50, а гораздо раньше — с 30 лет и делать колоноскопию раз в 6 или 12 месяцев, чтобы не пропустить развитие заболевания.
Выявление синдром Линча у пациентки с раком матки может повлиять и на лечение.
При поздних стадиях пациентам с синдромом Линча мы назначаем специфическую иммунотерапию препаратом пемпролизумаб, что позволяет улучшить прогнозы пациентов.
Генетическое профилирование опухоли – это колоссальный прорыв, который позволил нам подойти к полностью персонализированной терапии в онкологии, основанной не только на диагнозе, но и на понимании биологии опухоли. Для пациентов — это возможность получить точное узкоспециализированное лечение, дающее лучшие результаты, а в случае наследственных раков — возможность защитить будущие поколения от опасных заболеваний.
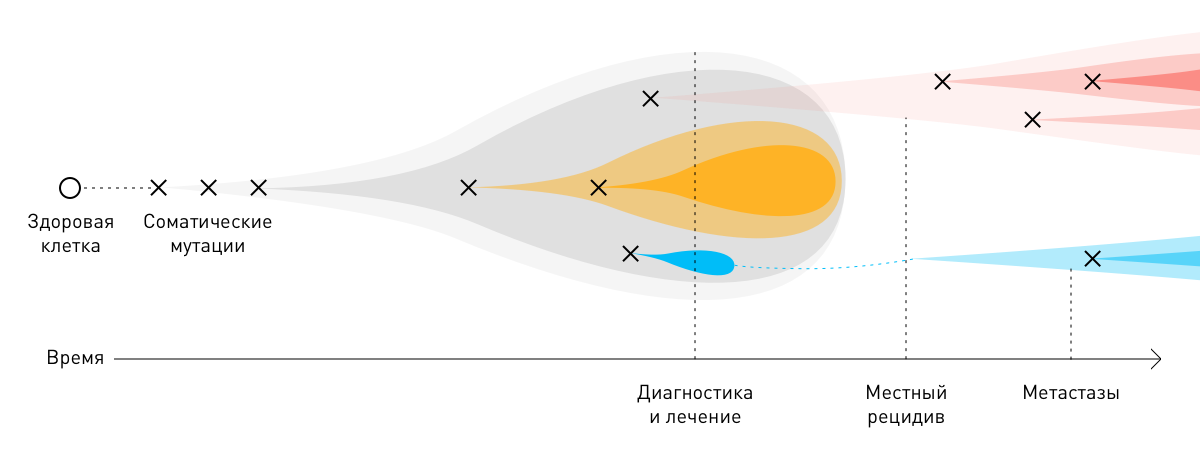
Мутация — любое изменение в молекуле ДНК либо структуре хромосом. Изменения, которые происходят в гаметах — половых клетках, называют герминальными, в любых других клетках организма — соматическими. Герминальные мутации передаются от родителей по наследству в виде генетических заболеваний. Соматические изменения не наследуются, а возникают в результате внешних воздействий (курения, радиоактивного излучения и других).
Доказано, что злокачественные опухоли в 90–95 % случаев возникают в результате соматических мутаций (спорадический рак). Наследственный рак, развивающийся в результате герминальных изменений, составляет 5–10 % от всех случаев заболевания.
Клеточные механизмы возникновения рака: протоонкогены и онкосупрессоры
Соматические мутации увеличивают шанс превращения здоровых клеток в раковые за счет активации онкогенов — генов, стимулирующих образование злокачественных опухолей. Онкогены образуются из обычных генов — протоонкогенов.
Деление клеток опухоли и разрастание опухолевой ткани тормозят антионкогены — гены-супрессоры. Протоонкогены и гены-супрессоры образуют систему стимуляции и подавления злокачественного процесса.
В подавляющем большинстве случаев раковые опухоли возникают из одной клетки в результате двух последовательных мутаций: первичной (герминальной) и вторичной (соматической). Для развития новообразования достаточно 3–6 таких повреждений. Вызванные ими изменения постепенно накапливаются в ДНК, вызывают неконтролируемое размножение пораженных клеток и образование атипичных тканей.
Драйверные соматические мутации и их использование для лечения злокачественных опухолей
В этиологии опухолей изменения в клетках вызывают ошибки копирования ДНК при делении — соматические драйверные мутации. В одних органах и тканях стволовые клетки делятся чаще, чем в других. В них злокачественные новообразования образуются чаще, чем в других структурах организма. По данным международного агентства по исследованию рака IARC, риск заболеть раком, а также выживаемость раковых клеток на 70–80 % зависят от интенсивности деления стволовых клеток и увеличиваются с возрастом.
Генетические маркеры опухолей (изменения в структуре белков в результате соматических драйверных мутаций) можно использовать как мишень и прицельно блокировать с помощью фармакологических препаратов. На этом принципе основан один из наиболее результативных современных методов лечения рака — таргетная терапия. Она позволяет:
- значительно повысить эффективность противоопухолевого лечения;
- снизить его токсичность и тем самым существенно улучшить состояние пациента.
Выявление драйверных соматических мутаций для диагностики злокачественных опухолей
Чем больше генетических маркеров известно, тем более адекватно подбирается лечение. Для выявления соматических мутаций при диагностике злокачественных опухолей используют следующие методики:
- секвенирование генома;
- выявление специфической аллели на амплифицированной ДНК;
- масс-спектрометрия;
- флуоресцентная гибридизация in situ (FISH).
Целесообразность использования того или иного метода в каждом конкретном случае определяет врач.
Соматические мутации опухолей: влияние на диагностику и лечение
В процессе роста злокачественные новообразования приспосабливаются к факторам онкологического лечения — лучевой, химио- и иммунотерапии. Молекулы онкологических клеток, выжившие после того или иного лечебного этапа, нередко мутируют. Соматические мутации опухолей способствуют прогрессированию болезни и её более агрессивному (рецидивирующему и метастатическому) течению. Из-за индивидуальных различий на молекулярном уровне общеклинические стратегии лечения того или иного вида рака могут быть неэффективны для отдельных пациентов.
Чтобы максимально снизить риск рецидивов, улучшить качество жизни больных и увеличить её продолжительность, соматические мутации в опухолях в ответ на фармако- и химиотерапию необходимо учитывать. Для их выявления разработан инновационный метод исследования — молекулярное профилирование. Результаты этого вида диагностики позволяют врачу назначать наиболее оптимальное персонализированное лечение.
Рак является генетическим заболеванием на клеточном уровне. Рак возникает результате накопления мутаций в генах, которые контролируют размножение клеток и их отношения друг с другом. Итак, чем же является то, что мы определяем как повреждение ДНК (мутация)?
Внутри в наших клеток находится основная молекула управления, ДНК. ДНК, с помощью содержащихся в ней генов, позволяет реализовать жизненно важные функции в нашем организме. ДНК представляет собой двухцепочечную структуру, состоящую из повторяющихся блоков, образованных в форме лестницы нуклеотидов. Многие факторы с которыми мы сталкиваемся в нашей повседневной жизни, такие как курение, некоторые химические вещества, инфекционные агенты и ультрафиолетовые лучи, могут нарушить структуру ДНК и разорвать взаимные последовательности нуклеотидов. Когда данное событие происходит в ДНК, его называют повреждение ДНК или мутация.

Чтобы описать мутацию примером из нашей из повседневной жизни; представим, что вся наша генетическая информация (геном), это книга. Алфавитом в этой книге являются 4 нуклеотида, которые формируют структуру ДНК, а каждый заголовок отображает наши гены. Если из книги удалить какую нибудь букву или вырвать какую нибудь страницу, она начнет становится бессмысленной. Точно также происходит и повреждение генов в организме человека.
Наш организм разработал ряд систем обороны против повреждения ДНК. Эти системы называются системами репарации ДНК. Мутации, происходящие в генах данной системы, являются одной из главных причин возникновения рака. Поврежденные в результате лечения гены раковых клеток, ремонтируются посредством данной системы. Одним из актуальных направлений исследований в области рака в последние годы, является разработка методов лечения, нацеленных на системы репарации ДНК, таких как (Олапариб и Нирапариб.

a) Мутация в зародышевой линии: Мутации, возникающие в репродуктивных клетках (гаметы), переходят потомству. В этом случае, мутации происходят в каждой клетке организма. Причиной наследственных синдромов рака являются поврежденные гены, которые передаются потомству. Например мутации происходящие в генах BRCA1 и BRCA2 в зародышевой линии, повышают риск наследственного рака груди или яичников. Наследственный рак составляет 10% всех случаев рака.
Более чем в 50% раковых заболеваний человека, присутствует повреждение гена P53. Мутация зародышевой линии, редко наблюдается в гене P53. Поскольку ген P53 представляет собой ген управления, он способствует началу образования рака, если в результате повреждения, начинается неконтролируемая работа этого гена, то остальные гены также перестают нормально функционировать.
b) Соматическая мутация: Повреждения, возникающие в клетках тела. Эти повреждения потомству не переходят. Повреждения, происходящие в генах, остаются в организме человека на протяжении всей его жизни. Образ жизни (Ожирение, солнечное воздействие, курение, инфекции) являются наиболее важным фактором в возникновении генных повреждений. Такие факторы, как курение, ультрафиолетовое излучение (УФ), вирусы и возраст, вызывают повреждение гена в клетках. Рак, возникающий в результате повреждения гена в клетках тела, называется спорадическим раком. Они составляют 80 процентов всех случаев рака.
Для того, чтобы объяснить причину возникновения рака, повреждение ДНК (мутация), имеет важное место в исследованиях рака. Следует подчеркнуть, что многие виды рака связаны не только с одним геном. Рак возникает в результате отношений нескольких генов друг с другом и с окружающей средой. В последние годы, более глубокое понимание повреждений ДНК стало возможным благодаря эпигенетической науке, которая изучает взаимосвязь между генами и окружающей средой.
1. Krishna L. Kanchi.
Integrated analysis of germline and somatic variants in ovarian cancer.
Nature Communications 5, Article number: 3156 (2014)
2. Greenman, Christopher et al.
“Patterns of Somatic Mutation in Human Cancer Genomes.”
Nature 446.7132 (2007): 153–158. PMC. Web. 4 Nov. 2016
Чем больше в раковой клетке мутаций, тем проще иммунитету её поймать.
Разные раковые опухоли по-разному реагируют на лечение, даже если речь идёт о какой-то продвинутой иммунотерапии, вроде той, за которую в прошлом году дали Нобелевскую премию. В каких-то случаях опухолевые клетки удаётся довольно быстро истребить, в других случаях, наоборот, опухоль оказывается очень устойчива к терапии.
Методы лечения в онкологии чреваты побочными эффектами, и эффекты эти в случае иммунотерапии могут быть особенно чувствительны (потому что простимулированный иммунитет начинает атаковать не только больные клетки, но и здоровые). И понятно, почему исследователи всячески стараются найти способ заранее угадать, как опухоль будет реагировать на тот или иной способ лечения: если заранее будет известно, что, например, иммунотерапия не даст ничего, кроме побочных эффектов, то и не стоит её пробовать.
Иммунитет узнаёт злокачественные клетки потому, что они отличаются от здоровых, а отличаются они потому, что в них есть мутации. То есть, с одной стороны, именно из-за мутаций клетка начинает бесконтрольно делиться, но именно они делают её видимой для иммунных охотников. Причём генетических дефектов в раковых клетках бывает не одна, не две, а множество.
Можно предположить, что чем больше мутаций, тем сильнее клетка будет выделяться среди здоровых, и тем проще иммунной системе её обнаружить. Поскольку иммунотерапевтические методы обычно сводятся к тому, чтобы простимулировать иммунитет человека против опухоли, то, вероятно, такая терапия будет наиболее эффективной против опухолей с наибольшим числом мутаций.
Исследователи из Мемориального онкологического центра им. Слоуна–Кеттеринга проанализировали ДНК достаточно развитых опухолей более чем у 1600 пациентов, которым действовали на иммунные контрольные точки (то есть использовали тот самый нобелевский метод лечения, когда иммунитету, грубо говоря, отключают тормоза). Кроме того, опухолевую ДНК анализировали ещё более чем у 5300 больных, к которым эту терапию не применяли, но которые лечились другими способами. Виды опухолей были самые разные, от меланомы до рака груди.
Похожие исследования выполняли и раньше, но, как пишет портал Nature, на этот раз в работе использовали очень широкий спектр опухолей, которые к тому же брали у больных после разных видов терапии. В статье в Nature Genetics говорится, что лучше всего иммунная терапия действительно срабатывала с теми опухолями, в которых было больше всего мутаций. Но для разных видов рака это пороговое количество мутаций было разным. То есть терапию, в которой действуют на иммунные контрольные точки и отключают иммунные тормоза, нужно соотносить не с абстрактным числом мутаций, но ещё и с конкретным видом опухоли.
Правда, как мы знаем, мутация мутации рознь, не все они одинаковы и не все делают раковую клетку более видимой для иммунной системы. Поэтому, если говорить о клинических перспективах, то, наверно, чтобы мы точно могли угадать реакцию опухоли на лечение, нужно знать не только число мутаций, но и что они в раковой клетке делают.
Рак груди представляет собой одно из самых часто встречающихся онкологических заболеваний среди женщин, совокупный риск которого, по современным оценкам, составит к возрасту 85 лет для девочек, родившихся в 1990 г., около 12,6% (иначе говоря, заболеть может 1 из 8 девочек). Предположение о существовании гена (генов), ответственного за наследственную предрасположенность к раку груди, впервые было высказано более 100 лет назад. Когда оно было подтверждено, то оказалось, что примерно 5—10% всех случаев рака груди контролируются мутациями определенных генов (к настоящему моменту были' картированы два таких гена — по одному на хромосомах 17 и 13).
Мутации, т.е. изменения наследственного аппарата клетки, затрагивающие целые хромосомы или их части, — наиболее часто встречающиеся примеры механизмов неменделевской генетики. Рассмотрим кратко одну из классификаций мутаций, разделяющую два их типа: гаметные (генеративные) и соматические. Первые изменяют гены, находящиеся в половых клетках; вторые — в клетках тела.
Гаметные мутации не влияют на фенотип родителей, поскольку они происходят во время формирования гаметы, т.е. когда фенотип родителя уже сформировался. Но с момента возникновения новой мутации она передается из поколения в поколение по законам Менделя. В результате таких мутаций, возникающих в поколении Fo (поколение родителей), фенотипически не проявляющих признаков болезни, а затем передающихся из поколения Fl в последующие поколения (F2, F3, . Fn) по законам Менделя, развиваются многие наследственные заболевания. Если мутация не летальна и не ведет к серьезному повреждению репродуктивной способности, процесс передачи мутировавшего гена из поколения в поколение приводит к появлению родословных со многими носителями мутации, начавшейся только в одном аллеле (на одной из хромосом представителя поколения Fo). Так, одна из мутаций гена на хромосоме 17, приводящая к развитию раковых заболеваний, вызывает примерно 57% всех наследуемых случаев рака груди. Механизм возникновения вредоносных мутаций неизвестен. Предполагается, что в большинстве случаев это спонтанные мутации. Не установлено также, происходят они в одном аллеле (у одного индивидуума) и затем распространяются в популяции или одинаковые мутации происходят у нескольких индивидуумов.
Конец страницы №82
Начало страницы №83
До сих пор мы говорили о гаметных мутациях. Однако примерно
случаев заболевания рака груди развивается в результате возникновения соматических мутаций.
Индуцированные мутации. До сих пор речь шла о спонтанных мутациях, т.е. происходящих без какой-либо известной причины. Возникновение мутаций — процесс вероятностный, и, соответственно, существует набор факторов, которые на эти вероятности влияют и изменяют их. Факторы, вызывающие мутации, называются мутагенами, а процесс изменения вероятностей появления мутации — индуцированием. Мутации, возникающие под влиянием мутагенов, называют индуцированными мутациями.
В современном технологически сложном обществе люди подвергаются воздействию самых разных мутагенов, поэтому изучение индуцированных мутаций приобретает все большее значение.
К физическим мутагенам относятся все виды ионизирующих излучений (гамма- и рентгеновские лучи, протоны, нейтроны и др.), ультрафиолетовое излучение, высокие и низкие температуры; к химическим — многие алкилирующие соединения, аналоги азотистых оснований нуклеиновых кислот, некоторые биополимеры (например, чужеродные ДНК и РНК), алкалоиды и многие другие химические агенты. Некоторые мутагены увеличивают частоту мутаций в сотни раз.
К числу наиболее изученных мутагенов относятся радиация высоких энергий и некоторые химические вещества. Радиация вызывает такие изменения в геноме человека, как хромосомные аберрации и потерю нуклеотидных оснований (гл. IV). Частота встречаемости мутаций половых клеток, индуцированных радиацией, зависит от пола и стадии развития половых клеток. Незрелые половые клетки мутируют чаще, чем зрелые; женские половые клетки — реже, чем мужские. Кроме того, частота мутаций, индуцированных радиацией, зависит от условий и дозы облучения.
Соматические мутации, возникающие в результате радиации, представляют собой основную угрозу населению, поскольку часто появле-
Конец страницы №83
Начало страницы №84
ние таких мутаций служит первым шагом на пути образования раковых опухолей. Так, одно из наиболее драматических последствий Чернобыльской аварии связано с возрастанием частоты встречаемости разных типов онкологических заболеваний. Например, в Гомельской области было обнаружено резкое увеличение числа детей, больных раком щитовидной железы. По некоторым данным, частота этого заболевания сегодня по сравнению с доаварийной ситуацией увеличилась в 20 раз.
ЭКСПАНСИЯ (ИНСЕРЦИЯ) ПОВТОРЯЮЩИХСЯ НУКЛЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ: МИОТОНИЧЕСКАЯ ДИСТРОФИЯ (МД)
Встречаемость миотонической дистрофии составляет 1 на 8000. Это заболевание наследуется как аутосомное доминантное заболевание и представляет собой наиболее часто встречающуюся форму мышечной дистрофии у взрослых. Клинически это заболевание крайне разнообразно; его симптомы включают: миотонию, прогрессирующую слабость, атрофию мышц, расстройства сердечно-дыхательной системы, катаракты, раннее облысение, умственную отсталость и атрофию половых органов. Обычно первые клинические проявления МД наблюдаются в 30—40 лет, однако в некоторых случаях она развивается с момента рождения, и тогда ее симптоматика намного тяжелее. Врожденная МД отличается высокой смертностью, у выживших же детей классическая симптоматика МД обнаруживается уже к 10-летнему возрасту.
Мутация, вызывающая развитие МД, была выявлена, описана и картирована. Биологический механизм этой мутации связан с нестабильной природой повторяющейся последовательности азотистых оснований (о структуре ДНК — гл. IV) на участке гена, расположенном на длинном плече хромосомы 19 (гл. I). Генетический механизм нестабильных повторяющихся последовательностей был открыт сравнительно недавно. По неизвестной до сих пор причине короткие сегменты ДНК, состоящие из 2, 3 и 4 нуклеотидов (гл. I), выстраивают повторяющиеся последовательности, которые включают от двух до нескольких сотен таких сегментов. Повторяющуюся последовательность можно представить следующим образом:
АСАСТ — сегмент повторяющейся последовательности;
АСАСТ АСАСТ АСАСТ АСАСТ АСАСТ— повторяющаяся последовательность из 5 сегментов;
(А) АСАСТ АСАСТ, (а) АСАСТ АСАСТ АСАСТ АСАСТ - 2 разных аллеля (А и а) локуса, содержащего повторяющуюся последовательность. На языке генетики это означает, аллель А содержит 2 повтора (2 сегмента нуклеотидов), а аллель а содержит 4 повтора (4 сегмента нуклеотидов).
Конец страницы №84
Начало страницы №85
Сегодня эти повторяющиеся последовательности найдены более чем в 50 000 локусов человеческого генома. Каждый локус содержит несколько (иногда до 20 и более) аллелей, включающих разное количество таких повторяющихся последовательностей. Эти аллели обычно наследуются по законам Менделя, однако были обнаружены и отклоняющиеся от них случаи, когда при переходе от одного поколения к другому количество повторяющихся сегментов меняется. Благодаря этому, а также высокой вариативности аллелей в каждом локу-се, повторяющиеся последовательности привлекают особое внимание генетиков, занимающихся картированием и локализацией генов в геноме человека.
Было замечено, что чем больше количество повторяющихся последовательностей (т.е. чем длиннее вся повторяющаяся последовательность) у больных с МД, тем тяжелее протекает заболевание (табл. 3.1).
Фенотипические проявления МД в зависимости от количества сегментов нуклеотидных повторяющихся последовательностей
| Фенотип | Клинические | Количество повто- |
| симптомы | ряющихся сегментов | |
| Легкая форма | катаракты | 50-150 |
| МД | ||
| Классическая | миотония | 100-1000 |
| форма МД | мышечная атрофия | |
| преждевременное облысение | ||
| атрофия половых органов | ||
| кардиорасстройства | ||
| Врожденная МД | гипотония | > 1000 |
| умственная отсталость | ||
| дисплегия |
Конец страницы №85
Начало страницы №86
6.НАСЛЕДОВАНИЕ СЛОЖНЫХ ПОВЕДЕНЧЕСКИХ ПРИЗНАКОВ
Большинство признаков, изучаемых психогенетикой, характеризуется тем, что в середине вариационного ряда (ряда значений) такого признака располагаются одна или две максимальные частоты, а справа и слева от них располагаются убывающие к концам ряда частоты, причем правые и левые частоты, одинаково удаленные от среднего, примерно равны. Оно относится к классу количественных и имеет нормальное (или приближающееся к нему) распределение. Его свойства описаны в любом руководстве по статистике, поэтому излагать их здесь мы не будем. Отметим только, что кривая нормального распределения имеет чрезвычайно важное значение для психологии. Дело в том, что каждый психологический признак в своем развитии зависит от очень большого количества факторов (и многих генов, и многих средовых обстоятельств), действующих в благоприятном или неблагоприятном направлении. И именно нормальное распределение отражает фенотипическое разнообразие, возникающее в результате воздействия множественных факторов на исследуемый признак.
Предваряя изложение того, что известно о наследовании количественных признаков, приведем более развернутый пример психогенетических исследований изменчивости сложных фенотипов человека.
Конец страницы №86
Начало страницы №87
интеллекту обычно имеют детей, чьи интеллектуальные способности выше среднего (об ограничениях, связанных с такой интерпретацией семейных данных, — в гл. VI, VII, VIII). Однако механизм передачи по наследству интеллектуальных способностей не соответствует законам Менделя.
Корреляции, описывающие сходство родственников по тестам интеллекта, также зависят от степени их кровного родства. Только супруги, в отличие от других неродственников, коррелируют между собой по интеллекту с коэффициентом г = 0,30-0,40. Это — весьма примечательная находка, имеющая особое значение для интерпретации сиблинговых и близнецовых корреляций (подробнее — в гл. VII, IX).
Что же известно сегодня о механизмах передачи по наследству сложных поведенческих, т.е. количественных, континуальных, признаков? Каковы генетические модели, описывающие механизмы этой передачи?
Наследуемость сложных поведенческих признаков обеспечивается, очевидно, многими генами. Такие признаки называются полигенными.
Когда законы Менделя были переоткрыты в начале XX столетия, серьезные теоретические баталии развернулись между теми, кто считал, что все признаки наследуются согласно закону Менделя (так называемые мендели-
Конец страницы №87
Начало страницы №88
сты), и теми, кто утверждал, что законы Менделя неверны (так называемые биометристы), поскольку они не могут объяснить передачу по наследству сложных поведенческих признаков.
Как выяснилось, обе стороны были одновременно и правы, и неправы. Менделисты были правы в том, что признаки, контролируемые одним геном, наследуются по законам Менделя. Ошибочность их позиции заключалась в том, что они считали законы Менделя универсальными, применимыми ко всем, в том числе сложным, признакам. Биометристы были правы в том, что многие сложные признаки распределены непрерывно, а не альтернативно, но заблуждались, утверждая, что законы Менделя справедливы только для растений.
Противоречие этих двух позиций разрешилось само собой, когда выяснилось, что законы Менделя применимы к наследованию полигенных признаков при отдельном рассмотрении каждого из генов, вовлеченных в генетический контроль исследуемого признака.
Кроме того, на функционирование каждого гена оказывают влияние характеристики среды. Предположим, что некоторый ген А чувствителен к изменению температуры в окружающей его клеточной среде (т.е. экспрессия гена зависит от характеристик окружающей среды). Тогда можно предположить существование следующей причинной цепочки: температура клеточной среды повышается (в ответ на какие-то внешние средовые воздействия или на внутреннюю реакцию организма, например, на инфекцию); в измененных температурных условиях аллель А2 производит больше белка (вероятнее всего, в какой-то своей измененной форме), который оказывает влияние на изучаемый нами признак, и признак проявляется сильнее. Рассуждение о подобных цепочках событий привело к возникновению еще одной модели наследования, называемой мультифакторной. Согласно этой модели, формирование признака контролируется сложным взаимодействием многих и генных, и средовых факторов.
Итак, в ситуации, когда рассматриваемый признак чувствителен к средовым влияниям, когда аллелей у каждого гена больше двух и когда каждый из этих аллелей может иметь или не иметь отличные по величине вклады в фенотип, все эти факторы приводят к формированию континуальных (непрерывных) распределений. Поэтому не удивительно, что часто в природе наблюдается континуальность, даже в тех случаях, когда сами аллели генов, контролирующих исследуемый признак, наследуются в соответствии с законами Менделя.
Представление о том, что количественные признаки формируются в результате действия множества генов, является краеугольным в разделе генетики, называемом генетикой количественных признаков. Эта область науки была разработана Р. Фишером и С. Райтом. Генетика количественных признаков представляет собой основу для общей теории происхождения (этиологии) индивидуальных различий, будучи междисциплинарной наукой. Ее междисциплинарность определяется как знаниями, создающими ее основу (общая биология
Конец страницы №88
Начало страницы №89
Основные понятия генетики количественных признаков представлены в гл. VI, VIII. Не останавливаясь здесь подробно на генных моделях, лежащих в основе генетики количественных признаков, укажем только, что полигенная модель, приведенная и обсуждаемая в этих главах, является базой для объяснения сходства родственников разной степени по фенотипическим признакам. Если генетические факторы влияют на формирование индивидуальных различий по какому-то признаку, то степень фенотипического сходства родственников должна изменяться в зависимости от степени их генетического родства (подробнее о разных методах психогенетики — в гл. VII, VIII). Например, родственники первой степени родства (родители — дети и родные сиблинги) в среднем имеют 50% общих генов. Иными словами, ребенок наследует примерно по 50% генов от каждого из родителей (но это — средняя величина; в каждом конкретном случае может быть и больше, и меньше). Если один из сиблингов унаследует какой-то аллель от одного родителя, то вероятность наследования того же аллеля другим сиблингом составит в среднем 50%.
В случае познавательных способностей (и некоторых заболеваний, например, шизофрении) степень фенотипического сходства между родственниками увеличивается по мере увеличения их генетической близости. Например, вероятность того, что отдельно взятый в популяции человек заболеет шизофренией, составляет 1%. Если же в семье есть больной, то риск заболевания шизофренией для его родственников второй степени (внуков и племянников) составит примерно 4%. Однако для родственников первой степени родства (родителей, сиблингов, детей) этот риск увеличивается до 9%. Наконец, риск развития шизофрении стремительно возрастает до 48% для монозиготных близнецов-шизофреников. Эта цифра намного больше цифры, полученной для дизиготных пар (17%).
Конец страницы №89
Начало страницы №90
Сходство родственников по анализируемым признакам позволяет утверждать, что генетические факторы влияют на количественные признаки, примером которых может служить как патология (например, шизофрения), так и норма (например, когнитивные способнос-
Конец страницы №90
Начало страницы №91
ти). Однако неоспоримым доказательством генетической этиологии анализируемых признаков сходство родственников считаться не может. Дело в том, что большинство пар родственников живут под одной крышей и проводят вместе много времени. Это сходство семейной среды также может играть существенную роль в формировании сходства родственников по фенотипическим признакам. Для того чтобы разделить вклады среды и генов, исследователи применяют специальные статистические модели или изучают несколько типов родственников одновременно (см. гл. VI—VIII).
Многие генетически контролируемые заболевания и поведенческие признаки развиваются в результате действия генетических механизмов, не подчиняющихся законам Менделя. Среди таких механизмов обычно называют хромосомные аберрации, сцепленное с полом наследование, импринтинг, появление новых мутаций, экспансии повторяющихся нуклеотидных последовательностей, наследование сложных континуально распределенных поведенческих признаков. Накопление новой информации, касающейся наследования немен-делирующих признаков, не опровергло законы Менделя. Выяснилось, что противоречия снимаются, когда все наследуемые признаки делятся на моногенные, в развитие которых вовлечен только один ген, и полигенные, в развитие которых вовлечено множество генов. Мен-делевские принципы сохраняют свою значимость при наследовании моногенных признаков, а также при наследовании каждого отдельно взятого гена, включенного в полигенную систему.
Конец страницы №91
Начало страницы №92
Г л а в а IV
Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций.
Поперечные профили набережных и береговой полосы: На городских территориях берегоукрепление проектируют с учетом технических и экономических требований, но особое значение придают эстетическим.

Общие условия выбора системы дренажа: Система дренажа выбирается в зависимости от характера защищаемого.
Читайте также: