
Острый лимфобластный лейкоз степень дифференцировки лейкозных клеток
К острым лимфобластным лейкозам (ОЛЛ) относятся лейкозы, опухолевый субстрат которых представлен клетками-предшественницами лимфоидного ряда. Выделяют два варианта лейкозов в зависимости от направленности дифференцировки бластных клеток: острый В-лимфобластный лейкоз/лимфома из клеток-предшественниц и острый Т-лимфобластный лейкоз/лимфома из клеток-предшественниц.
Большинство наблюдений расценивается как острый лейкоз, однако случаи с выраженным экстрамедуллярным поражением и числом бластов в костном мозге менее 25 %, по классификации ВОЗ относятся к лимфомам.
Наличие отдельных цитогенетических аномалий при остром лимфобластном лейкозе (ОЛЛ) имеет существенное прогностическое значение, поэтому при В-острый лимфобластный лейкоз (ОЛЛ) выделяется ряд вариантов в зависимости от характеристики кариотипа.
1. В-лимфобластный лейкоз/лимфома из предшественников: t(9;22)(q34;qll); BCR/ABL; t(v;llq23); MLL перестройка; t(l;19)(q23;pl3) PBX/E2A; t(12;21)(pl2;q22) TEL/АМН; ОЛЛ гиподиплоидный; ОЛЛ гипердиплоидный (>50).
2. Т-лимфобластный лейкоз/лимфома из предшественников.
Выделение морфологических типов лимфобластов по классификации FAB в зависимости от их размера [преобладание микроформ Л1 или мезоформ Л2] не сыграло существенной роли в определении тактики лечения или в прогнозе заболевания. Тип бластов с базофилией и вакуолизацией цитоплазмы ЛЗ более характерен для лимфомы Беркитта и неспецифичен для клеток-предшественниц. Морфоцитохимические признаки В- и Т-лимфобластов имеют некоторые различия:
• В-лимфобласты характеризуются значительным морфологическим разнообразием. Клетки могут быть правильной или вытянутой формы, иногда они напоминают очертания ручного зеркала. Размер бластов варьирует от микро- до мезоформ. Ядра округлые, овальные или складчатые, с плотным хроматином и редкими нуклеолами. Цитоплазма скудная, слабобазофильная, иногда содержит легкую зернистость. Ядерно-цитоплазматическое отношение высокое или умеренное.
• В-лимфобласты не содержат пероксидазы, липидов и ASD-хлорацетатэстеразы. В половине случаев в них обнаруживаются маленькие гранулы PAS-положительного вещества и кислая фосфатаза в мелкогранулярной форме.
• Т-лимфобласты, как правило, мономорфны, среднего размера, округлые, с ядрами неправильной складчатой формы. Цитоплазма скудная, умеренно базофильна. Ядерно-цитоплазматическое отношение высокое.
• Т-лимфобласты не содержат пероксидазы, ASD-хлорацетатэстеразы и липидов. PAS-положительное вещество обнаруживается редко в виде одной гранулы. Характерным признаком является наличие крупных гранул ферментов: кислой фосфатазы, неспецифической эстеразы и кислой неспецифической эстеразы.
В зависимости от стадии дифференцировки лимфобластов выделяются субварианты В- и Т-линейных острых лимфобластных лейкозов (ОЛЛ). Диагноз базируется на данных иммунофенотипирования клеток. Частота выявления этих субвариантов различна. Для разных субвариантов характерны различные хромосомные нарушения.
Лейкозы из В-линейных клеток-предшественниц встречаются приблизительно у 75 % взрослых больных острым лимфобластным лейкозом (ОЛЛ). Во всех случаях бластные клетки экспрессируют молекулы CD19 и/или CD79a и/или цито-плазматическую CD22. По мере дифференцировки к этим антигенам добавляются новые, специфичные для последующих этапов созревания. Среди В-линейных лейкозов выделяют следующие иммуноподварианты: про-В, пре-пре-В, пре-В и В-ОЛЛ. Наличие t(9;22), t(4;ll) и t(l;19), наблюдаемых при вариантах про-В и пре-пре-В, является независимым показателем неблагоприятного прогноза.
Транслокация (9; 22) встречается у взрослых больных острым лимфобластным лейкозом (ОЛЛ) достаточно часто (около 25 %). Ph-позитивные и Ph-негативные лимфобласты не имеют морфоцитохимических и иммунофенотипических различий, поэтому данные цитогенетического исследования являются необходимыми при выборе тактики лечения больных острым лимфобластным лейкозом (ОЛЛ). Прогностическое значение для субвариантов В-линейного острого лимфобластного лейкоза (ОЛЛ) имеет также изменение числа хромосом в бластах: гипер-плоидия является благоприятным, а гипоплоидия — неблагоприятным признаком.
По патогенетическому принципу, исходя из особенностей морфологической характеристики лейкозных клеток, лейкозы подразделяют на острые и хронические. К острым лейкозам относят опухоли с полной остановкой дифференцировки родоначальных кроветворных клеток на каком-то уровне созревания;субстрат опухоли составляют клетки II, ΙΙΙ и IV классов по современной схеме кроветворения. В группу хронических лейкозов входят опухоли с частичной задержкой созревания клеток и накоплением клеток определенной степени зрелости.
Острые лейкозы.Гематологическая картина в развернутой стадии заболевания характеризуется классической триадой:
· изменением содержания лейкоцитов (общее число лейкоцитов снижено, повышено или остается нормальным);
· появлением в крови большого числа бластных клеток;
Уже на ранних стадиях болезни отмечаются нормохромная анемия и тромбоцитопения, развитие которых обусловлено угнетением нормального гемопоэза вследствие продукции лейкозным клоном цитокинов, угнетающих пролиферацию нормальных стволовых клеток.
Составляющие субстрат опухоли бластные клетки при различных вариантах острого лейкоза морфологически трудно различимы, но могут быть дифференцированы с помощью цитохимических методов по разнице в содержании ферментов. Исходя из особенностей цитохимических свойств лейкозных клеток, острые лейкозы (упрощенно) подразделяют на:
Выделенные нозологические формы различаются также по клиническим признакам и, что особенно важно, по ответу на цитостатическую медикаментозную терапию.
Окончательный диагноз острого лейкоза (особенно в тех случаях, когда лейкемические клетки не выходят в периферическую кровь) должен ставиться на основании исследования пунктата костного мозга. При этом основным диагностическим признаком является мономорфная картина костного мозга с преобладанием однотипных бластных клеток. Морфологические критерии последних очень изменчивы; как и все опухолевые клетки, лейкемические бласты атипичны, отличаются прогрессирующей анаплазией. По мере прогрессирования заболевания вследствие опухолевой прогрессии и под влиянием цитостатической терапии бластные клетки могут до неузнаваемости изменять свою морфологию, утрачивать ферментную специфичность.
Хронические лейкозы.Несколько упрощенно классификацию хронических лейкозов можно представить в следующем виде:
Патогенез лейкозов
Согласно мутационно-клоновой теории происхождения лейкозов лейкозогенный фактор (ионизирующая радиация, химическое вещество, вирус и др.) вызывает мутацию (повреждение ДНК, нарушение генетического кода) одной из клеток-предшественниц гемопоэза ΙΙ-Ш классов. В результате нарушается информация деления и дифференцировки клеток, наблюдается выход их из-под контроля регулирующих систем организма. Это приводит к безудержному размножению определенной разновидности клеток. Таким образом, составляющие субстрат опухоли лейкозные клетки представляют собой моноклональное потомство первоначально мутировавшей клетки и сохраняют все присущие ей признаки. В моноклоновой стадии опухолевые клетки чувствительны к химиотерапии.
Убедительным подтверждением клонового происхождения лейкозов является обнаружение у подавляющего большинства людей, больных хроническим миелолейкозом (в 80-90% случаев) аномальной (с укороченным длинным плечом) так называемой филадельфийской (Ph') хромосомы во всех миелоидных клетках, включая гранулоцитарный, эритроидный и мегакариоцитарный ростки, возможно за исключением Т-лимфоцитов. Этот факт является неоспоримым доказательством происхождения хронического миелолейкоза из одного патологического клона, родоначальницей которого является плюрипотентная стволовая клетка-предшественница миелопоэза (КОЕ-ГЭММ).
В процессе развития лейкоза (опухолевая прогрессия)происходят качественные изменения составляющих субстрат опухоли клеток, обусловленные нестабильностью их генетического аппарата. Изменение генетической программы клетки по одному или нескольким свойствам наследуется дочерними клетками. Нарастающая опухолевая прогрессия создает неравнозначные условия для всех дочерних клеток. Часть из них лучше приспосабливается к меняющимся условиям, они интенсивно размножаются, передавая новые свойства своим потомкам. Лейкозы превращаются из моноклоновых в поликлоновые, причем преобладают менее дифференцированные и более злокачественные клеточные элементы.
В процессе опухолевой прогрессии формируется атипизм гемобластозов. Появляются существенные отличия бластоматозных клеток (в обменных процессах, энергетике, росте, структуре и функции) от нормально функционирующих. Трансформированный костный мозг начинает интенсивно продуцировать лейкозные клетки разной степени зрелости. По мере усиления пролиферативных процессов, нарастания массы опухолевой ткани в кровь начинает выбрасываться все большее количество лейкоцитов, их бластных, незрелых форм. Повышается содержание молодых форм гранулоцитарных клеток – базофилов, эозинофилов и особенно нейтрофилов, лимфобластов, пролимфоцитов. Функциональная активность этих клеток извращена или полностью подавлена со всеми вытекающими отсюда последствиями.
Отдельные клоны опухолевых клеток выходят из-под контроля регулирующих систем организма, становятся устойчивыми к проводящейся цитостатической терапии, метастазируют в органы и ткани, в норме в гемопоэзе не участвующие, образуя очаги экстрамедуллярного кроветворения.
| | | следующая лекция ==> | |
| Типовые формы патологии системы лейкоцитов | | | Локальная форма уравнений |
Дата добавления: 2018-06-28 ; просмотров: 487 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Острые миелоидные лейкозы
Острые миелоидные лейкозы (ОМЛ) представляют гетерогенную группу клональных заболеваний, при которых неопластическая трансформация наблюдается в мультилинейной стволовой клетке или в стволовой клетке с линейной дифференцировкой.
Поскольку мультипотентная стволовая клетка дает начало гранулоцитам, моноцитам, эритроцитам и мегакариоцитам, при ОМЛ возможно поражение всех или отдельных клеточных линий.
В целом неопластический клон прекращает дифференцировку на стадии бласта, что ведет к прогрессирующему накоплению бластов в костном мозге (КМ) с последующим их выходом в периферическую кровь (Рис. 1, 2).

Рис. 1. Схема кроветворения

Рис. 2. Мезенгиогенез. Мезенхимальная стволовая клетка (МСК)
ОМЛ составляют около 20% от всех лейкозов и диагностируются в любом возрасте, однако частота их возникновения увеличивается с возрастом, медиана возраста составляет 60-65 лет.
Заболеваемость составляет 2-3 человека на 100000 населения в год. Этиология острого миелоидного лейкоза неизвестна, однако причинными факторами могут быть: алкилирующие препараты, некоторые химические вещества, пестициды, рентгенконтрастные вещества, красители, миелотоксические агенты, ионизирующее излучение.
Дозы, превышающие 100 рад, оказывают дозо-зависимый эффект в отношении возникновения ОМЛ. Генетическая аномалия - трисомия Хр21 (синдром Дауна) вызывает повышенную частоту развития ОМЛ, хотя в большинстве случаев при этом синдроме развивается острый лимфобластный лейкоз (ОЛЛ).
Повышен риск развития ОМЛ при синдроме Блума, анемии Фанкони, атаксии-телеангиэктазии, нейрофиброматозе Реклингаузена и т.п. Гематологические нарушения - апластическая анемия, хронический миелопролиферативный синдром, миелодиспластические состояния, пароксизмальная ночная гемоглобинурия повышают риск развития острого миелоидного лейкоза.
Клинические проявления неспецифичны. Жалобы на общую слабость, недомогание, головокружение могут задолго предшествовать диагнозу. Нередко отмечается бледность кожных покровов, повышение температуры тела без наличия инфекции, довольно часто отмечается геморрагический синдром по петехиально-пятнистому типу, иногда кровотечения.
Оссалгии отмечаются у 20% больных. Гепатоспленомегалия не является диагностическим признаком ОМЛ, но отмечается у 50% больных. Лейкемиды кожи и инфильтрация десен обычно характерны для миеломоно- и монобластного варианта. Исходное поражение ЦНС встречается редко - при гиперлейкоцитозе и/или монобластном варианте ОМЛ.
Цитохимическими маркерами бластов гранулоцитарного ряда являются липиды, миелопероксидаза (МПО), ASD-хлорацетатэстераза (ASD-ХАЭ). Определение последнего фермента имеет небольшую диагностическую ценность вследствие невысокой ее активности. В диагностике М5а и М5в вариантов ОМЛ главную роль играет исследование неспецифической эстеразы (НЭ), подавляемой фторидом натрия (NaF), антилизоцима.
Применение иммунофенотипирования для диагностики острого миелоидного лейкоза позволяет определить линейность и/или этап дифференцировки бластов, начиная с уровня стволовых клеток-предшественников. Изучение кариотипа бластных клеток позволило выявить закономерные изменения, характерные для отдельных вариантов острых лейкозов.
При цитогенетическом исследовании у 70-80% больных ОМЛ можно выявить неслучайные приобретенные хромосомные аберрации. Некоторые из них, при которых образуется химерный сливной белок, могут служить маркером наличия опухолевого клона. Данные цитогенетического исследования используются для уточнения отдельных вариантов ОМЛ, определения прогноза заболевания, а также для контроля качества и эффективности проводимой терапии.
Таблица 1. Характеристика бластов при ОМЛ (М.А.Френкель, 2001)

Согласно FAB-классификации, выделяют следующие варианты острого миелоидного лейкоза.
Острые миелобластные лейкозы (М0, М1, М2). Этот термин объединяет три подтипа заболевания, которые отличаются друг от друга по степени дифференцировки лейкемических клеток -миелобластов.
М0 составляет около 3-5% ОМЛ. Диагноз может быть установлен только при выполнении иммунофенотипирования. Бласты экспрессируют CD13, CD31. С помощью моноклональных антител (антимиелопероксидазы) - анти-МПО в бластах можно обнаружить фермент миелопероксидазу.
М1 составляет около 15% всех ОМЛ. Морфологически бласты имеют круглое или овальное ядро с рыхлым хроматином, несколько нуклеол и небольшой ободок серо-голубой цитоплазмы без гранул. Тельца Ауэра (первичные лизосомы) выявляются в 10% случаев. При этом варианте определяется минимальная степень миелоидной дифференцировки.
Иммунофенотипическими маркерами является выявление антигенов CD13, 33, 34, HLA-DR. В единичных клетках выявляется транслокация хромосом (Хр) (t 9; 22) и инверсия Хр3. МПО и/или липиды содержатся более чем в 3% бластов.
М2 составляет около 25-32% всех ОМЛ. Более 10% бластов содержит зернистость в цитоплазме в виде нежных темно-красных гранул. Бласты содержат МПО, липиды, гранулоцитарную эстеразу (ГЭ) в большинстве клеток, ШИК-вещество в диффузной форме; бласты экспрессируют антигены CD11, 13, 15, 33, анти-МПО. Примерно в 20-40% случаев выявляется t (8; 21); при этом отмечаются спленомегалия, хлоромы, эозинофилия в периферической крови.
При данной транслокации на длинном плече Хр8 образуется химерный ген AML1-ETO, продуктом деятельности которого является патологический белок CBFa-ETO. При связывании его с ДНК происходит ингибирование транскрипции и, соответственно, нарушаются механизмы дифференцировки миелоидных клеток.
М2 базофильно-клеточный представлен в единичных случаях. Бласты содержат грубую базофильную зернистость, не содержат миелопероксидаза, липиды и ГЭ. Цитогенетически в этих случаях может выявляться t (6; 9). В сыворотке крови повышено содержание гепарина и серотонина. При этой форме острого лейкоза прогноз крайне неблагоприятный.
М3 составляет около 10% случаев острого миелоидного лейкоза. Ядра бластов имеют характерную лопастную форму, цитоплазма содержит обильную азурофильную зернистость и палочки Ауэра. В зависимости от величины гранул выделяют макрогранулярный и микрогранулярный (M3v) варианты данного заболевания. Бласты содержат большое количество МПО, липидов, ГЭ, умеренное количество НЭ, неподавляемой NaF; ШИК-вещество в диффузной форме.
Поскольку в гранулах и тельцах Ауэра содержатся сульфатированные мукополисахариды, обладающие активностью типа тканевого тромбопластина, в большинстве случаев развивается ДВС-синдром (диссеминированное внутрисосудистое свертывание). Бласты экспрессируют антигены CD 11, 13, 15, 33, анти-МПО. В 1977 г. J.D.Rowley и соавторы установили, что в 95% случаев острого промиелоцитарного лейкоза (ОПЛ) обнаруживается t (15; 17).
Вследствие этого PML-ген (ген промиелоцитарного лейкоза), расположенный на Хр15, переносится на длинное плечо Хр17 в область, где находится ген а-рецептора ретиноевой кислоты (RARA). В норме этот ген участвует в регуляции дифференцировки миелоидных клеток. Ген PML является регулятором роста и играет роль в созревании и активации различных клеток.
Продуктом химерного гена PML-RARA является патологический белок, который накапливается в цитоплазме и ядре миелоидных клеток, что приводит к блоку дифференцировки клеток на уровне промиелоцитов. Кроме того, этот белок блокирует механизмы апоптоза, что поддерживает жизнеспособность опухолевых клеток.
В М4 варианте ОМЛ выделяют биклональный (1), бифенотипический (2) и миело-монобластный эозинофильный М4ЕО (3) подварианты. Биклональный вариант составляет около 17% всех случаев, при нем бласты представлены двумя типами клеток -миелобластами и монобластами, и экспрессируют антигены анти-МПО, анти-лизоцим, CD 13, 14, 15, 33. Цитогенетические исследования могут выявить t (8; 21).
Бифенотипический вариант крайне редок. Морфологически бласты характеризуются как миелобласты при М2 варианте, однако во всех бластах одновременно содержится МПО, ГЭ и НЭ, подавляемая NaF.
М5а составляет около 10% острого миелоидного лейкоза. Бласты не имеют специфических морфологических признаков, содержат НЭ, подавляемую фторидом NaF. Отдельные бласты содержат небольшое количество миелопероксидазы и/или липидов. Бласты экспрессируют антигены CD 13, 14, 15, 33, антилизоцим. Иногда (часто после проведенной полихимиотерапии) выявляется аномалия Хр11.
М5в встречается редко. Морфологически бласты имеют моноцитоидную форму ядер, содержат значительное количество НЭ, подавляемой NaF, небольшое количество МПО или липидов. Бласты экспрессируют антигены CD 13, 14, 15, 33, антилизоцим. Как и в предыдущем варианте, может выявляться аномалия Хр11.
Мб (1) - острый эритробластный лейкоз. Характеризуется наличием расширенного патологического красного ростка. Бласты имеют морфологические и цитохимические признаки миелобластов варианта М1. Клетки красного ряда имеют мегалобластоидные изменения с признаками морфологической и цитохимической дисплазии, содержат тельца Жолли, усилена сидерофилия.
Мб (2) - эритромиелоз. В костном мозге преобладает бластная популяция, которая состоит из миелобластов и эритробластов. Бласты не имеют специфических морфоцитохимических признаков, экспрессируют гликофорин А - эритроидный антиген. Цитогенетически можно выявить множественные хромосомные перестройки. Есть мнение, что изменения со стороны клеток красного ряда представляют реакцию на уже имеющийся ОМЛ. Цитогенетические исследования показывают наличие аномалий Хр5,7.
М7 встречается крайне редко. Мегакариобласты в большом количестве в костном мозге с их выходом в периферическую кровь. Цитохимически в бластах находят большое содержание а-нафтил-ацетатэстеразы, не ингибируемой NaF, и некоторое количество а-бутиратэстеразы, выявляется положительная ШИК-реакция. Бласты экспрессируют тромбоцитарные антигены CD 41 и/или CD 61.
Острые лимфобластные лейкозы
Острые лимфобластные лейкозы представляют гетерогенную группу клональных заболеваний лимфопоэтической ткани, характеризующуюся накоплением лимфобластов в КМ. Прогрессирующая инфильтрация опухолевыми лимфобластами костного мозга подавляет продукцию нормальных гемопоэтических клеток. ОЛЛ составляют 10% от всех лейкозов, причем 60% от этого количества наблюдается у детей.
В Европе ОЛЛ является наиболее частым опухолевым заболеванием детского возраста. Пик заболеваемости приходится на возраст 3-7 лет, второй, не очень выраженный пик, отмечается в возрасте 50-60 лет. Заболеваемость острым лимфобластным лейкозом ниже на Среднем Востоке и в Азии. В США частота заболеваемости белого населения в 2 раза выше, чем негроидного.
Подтип В-клеточного ОЛЛ составляет 80% всех случаев ОЛЛ, почти во всех остальных случаях диагностируется Т-ОЛЛ. Крайне редко определяется нуль-клеточный вариант.
Этиология ОЛЛ неизвестна. Не выявлено связи с миелотоксическими агентами, химикатами, ретровирусами, хотя внедрение ретровируса в геном при лечении агаммаглобулинемии Бруттона в некоторых случаях приводило к развитию острого лимфобластного лейкоза.
Умеренное повышение заболеваемости ОЛЛ отмечено под воздействием высоких доз ионизирующего излучения и при некоторых иммунодефицитных состояниях. Хотя вирус Эпштейн-Барра инкорпорируется в геном клеток при африканском варианте лимфомы Беркитта, вирусный геном отсутствует при лимфобластном L3 варианте ОЛЛ. Заболеваемость ОЛЛ в 15-20 раз выше при синдроме Дауна.
В патогенезе ОЛЛ основным является прогрессирующее накопление лимфобластов в КМ, что снижает потенциал дифференцировки и созревания. Для популяции лейкемических лимфобластов характерна остановка дифференцировки трансформированных лимфопоэтических стволовых клеток на специфической стадии, что снижает также уровень апоптоза.
Лейкемические лимфобласты имеют удлиненное время генерации (3 суток) в сравнении с нормальными лимфоидными предшественниками (1 сутки). Поскольку лимфобласты не созревают до более дифференцированной стадии, они накапливаются в костном мозге. Клинически это проявляется анемией, гранулоцитопенией и тромбоцитопенией.
Клиника ОЛЛ обычно представлена неспецифическими симптомами, хотя дебют может быть острым. Наиболее часто отмечаются слабость, сонливость, оссалгии, артралгии, не связанное с инфекцией повышение температуры тела. Иногда единственной жалобой являются оссалгии при отсутствии лимфоаденопатии, органомегалии и изменений в анализах периферической крови.
При вовлечении в процесс ЦНС отмечаются головная боль, тошнота, рвота. Бактериальная инфекция встречается при выраженной нейтропении. При физикальном обследовании может отмечаться бледность кожных покровов, геморрагический синдром по петехиально-пятнистому типу, лимфоаденопатия (чаще в области шеи), гепатоспленомегалия. Крайне редко бывают лейке -миды кожи.
В анализе крови в 60% случаев отмечается сублейкоцитоз, в 10% - гиперлейкоцитоз свыше 100,0х10 9 /л, тромбоцитопения менее 50,0х10 9 /л - у 60% больных в момент диагностики. При гиперлейкоцитозе почти всегда отмечается лимфоаденопатия, гепато- и спленомегалия. Анемия является непостоянным признаком.
В пунктате КМ отмечается гиперклеточность с наличием более 20% бластов. Для установления диагноза необходимо комплексное (морфологическое, цитохимическое, иммунофенотипическое, цитогенетическое) исследование бластных клеток.
Цитогенетическое изучение выявило некоторые аномалии количества хромосом или их структуры в 90% случаев острого лимфобластного лейкоза. Гипердиплоидность (47 или более хромосом) выявлена у 1/3 больных. Обычно существуют хромосомные транслокации, вовлекающие протоонкогены; большинство из них демонстрируют транслокацию c-myc протоонкогена Хр8 в регион гена Хр14, кодирующий иммуноглобулины (Ig). - t (8; 14).
В случаях В-ОЛЛ встречаются t (2; 8) или t (8; 22). Это специфические транслокации, т.к. локус с c-myc дерегулируется промотором Ig. При пре-В-клеточном варианте острого лимфобластного лейкоза специфическая транслокация t (1; 19) наблюдается в 25% случаев. В результате гибридный ген Хр19 кодирует белок, который нарушает регуляцию транскрипции.
Описаны транслокации при Т-клеточном остром лимфобластном лейкозе, вовлекающие Хр11 и локус Т-клеточного рецептора (TCR) на Хр14 и Хр7, а также делеции Хр6, Хр9, Хр12, так что в этих случаях может происходить потеря опухолевых генов-супрессоров. Филадельфийская хромосома (Ph), t (9; 22) выявляется в 20% ОЛЛ у взрослых и менее чем в 5% у детей. Патологический белок, кодируемый гибридным геном при этой транслокации, при ОЛЛ отличается от белка, продуцируемого химерным геном BCR/ABL при хроническом миелолейкозе.
Лимфобласты при ОЛЛ всегда весьма гетерогенны.
Выделяют три подтипа острого лимфобластного лейкоза:
L1 - лимфобласты малой величины, имеют круглое или овальное ядро с тонко организованным хроматином, единичными нуклеолами и ободком голубой цитоплазмы.
L2 - лимфобласты средней и большой величины, имеют ядро неправильной формы с рыхлым хроматином, одну или более нуклеол и ободок умеренно серо-голубой цитоплазмы.
L3 - лимфобласты крупных размеров, с круглым или овальным ядром, содержащим глыбчатый хроматин, 2-5 нуклеол и ободок темно-голубой и частично вакуолизированной цитоплазмы.
С учетом иммунологических маркеров можно выделить четыре подтипа ОЛЛ, три из них В-клеточного и один - Т-клеточного происхождения.
Имеются два подтипа, состоящие из предшественников В-клеток:
- пре-пре-В клеточный ОЛЛ (морфологически - L1 вариант),
- пре-В клеточный ОЛЛ (морфологически - L2 вариант).
Третий подтип В-клеточного ОЛЛ состоит из более зрелых В-клеток (L3 вариант).
Около 20% случаев ОЛЛ происходят из предшественников Т-клеток и относятся морфологически к L2 варианту. Демонстрируется реаранжировка генов в- или гамма-цепей рецептора Т-клеток (TCR).
ОЛЛ из нуль-клеток - это малая пропорция случаев ОЛЛ, отличающаяся по маркерам от В- или Т-линии лимфоцитов и поэтому называемая нуль-ОЛЛ или неклассифицируемым ОЛЛ.
При цитохимическом исследовании характерно наличие терминальной динуклеотидил-трансферазы (Тс1Т) в 95% случаев пре-пре-В-, пре-В клеточного ОЛЛ и Т-клеточном ОЛЛ.
Активность фермента снижена в зрелых В-лимфоцитах и при L3 варианте ОЛЛ. При ОНЛЛ активность фермента выявляется только в 5-10% случаев.
Таблица 2. Характеристика бластов при ОЛЛ (М.А. Френкель, 2001)

Реакции на МПО, специфические и неспецифические эстеразы, ШИК-реакция - отрицательные. Реакция на окрашивание масляным красным - положительная.
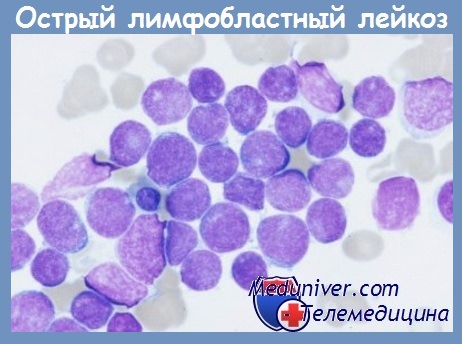
Лимфобластный лейкоз – злокачественное заболевание кроветворной системы, характеризующееся быстрым и неконтролируемым увеличением количества лимфобластов (незрелых лимфоидных клеток).
В педиатрической практике это наиболее часто встречающееся онкологическое заболевание. В общей структуре заболеваемости системы кроветворения у детей на долю острого лимфобластного лейкоза приходится 75-80%. Девочки болеют несколько реже мальчиков. Пик заболеваемости приходится на возраст от года до шести лет.

Взрослые люди заболевают лимфобластным лейкозом в 10 раз реже, чем дети. Показатель заболеваемости возрастает среди пациентов старше 60 лет.
У детей лимфобластный лейкоз обычно развивается как первичное заболевание, в то время как у взрослых пациентов он чаще возникает как осложнение хронического лимфоцитарного лейкоза.
При остром лимфобластном лейкозе у детей прогноз хороший. Современная полихимиотерапия позволяет добиться стойкой ремиссии у 95% пациентов этой возрастной группы.
Причины и факторы риска
К развитию острого лимфобластного лейкоза приводит образование в красном костном мозге злокачественных клонов, которые представляют собой группу кроветворных клеток, утративших способность контролировать размножение. К образованию клона приводят хромосомные аберрации:
- амплификация – образуются дополнительные копии определенного участка хромосомы;
- инверсия – поворот участка хромосомы;
- делеция – утрата участка хромосомы;
- транслокация – две хромосомы обмениваются между собой определенными участками.
Генетические аномалии, способствующие развитию лимфобластного лейкоза, возникают на этапе внутриутробного формирования плода. Однако для того чтобы запустился патологический процесс образования клеток-клонов необходимо воздействие провоцирующих внешних факторов. К таким факторам относятся:
Клиническое течение острого лимфобластного лейкоза у детей и взрослых стремительное. Нередко к моменту диагностирования заболевания масса всех лимфобластов в организме пациента достигает 3–5% от общей массы тела.
Формы заболевания
Лимфоциты – это разновидность агранулоцитных лейкоцитов, основными функциями которых являются:
- выработка антител (гуморальный иммунитет);
- непосредственное уничтожение чужеродных клеток (клеточный иммунитет);
- регуляция деятельности других типов клеток.
У взрослого человека лимфоциты составляют 25–40% от общего количества лейкоцитов. У детей их доля может достигать 50%.
Обеспечение регуляции гуморального иммунитета обеспечивают Т-лимфоциты. За стимуляцию выработки антител отвечают Т-хелперы, а за торможение – Т-супрессоры.
B-лимфоциты распознают антигены (чужеродные структуры) и вырабатывают против них специфические антитела.
NK-лимфоциты контролируют качество других клеток организма человека и активно уничтожают те из них, которые отличаются от нормальных (злокачественные клетки).
Процесс образования и дифференцирования лимфоцитов начинается с образования лимфобластов – лимфоидных клеток-предшественников. Из-за опухолевого процесса созревание лимфоцитов нарушается. В зависимости от типа поражения лимфоцитов лимфобластный лейкоз подразделяется на Т-линейный и В-линейный.
По классификации ВОЗ выделяют несколько типов острого лимфобластного лейкоза:
- пре-пре-В-клеточный;
- пре-В-клеточный;
- В-клеточный;
- Т-клеточный.
В общей структуре заболеваемости лимфобластными лейкозами на долю B-клеточных форм приходится 80-85%, а на Т-клеточных – 15-20%.
Стадии заболевания
В течении острого лимфобластного лейкоза выделяют следующие стадии:
- Начальная. Продолжается 1–3 месяца. В клинической картине преобладают неспецифические признаки (бледность кожи, субфебрильная температура, ухудшение аппетита, быстрая утомляемость, вялость). Некоторые пациенты жалуются на боли в мышцах, суставах и костях, животе, стойкие головные боли.
- Разгар. Ярко выраженные признаки заболевания, проявляющиеся анемическим, интоксикационным, гиперпластическим, геморрагическим и инфекционным синдромом.
- Ремиссия. Характеризуется нормализацией клинико-гематологических показателей.
- Терминальная стадия. Характерно быстрое прогрессирование симптомов лимфобластного лейкоза. Заканчивается летальным исходом.
Симптомы лимфобластного лейкоза
Клиническое течение острого лимфобластного лейкоза у детей и взрослых стремительное. Нередко к моменту диагностирования заболевания масса всех лимфобластов в организме пациента достигает 3–5% от общей массы тела. Это связано с бурной пролиферацией клеток-клонов.
У взрослых прогноз при лимфобластном лейкозе серьезен, пятилетняя выживаемость не превышает 34–40%.
В клинической картине лимфобластного лейкоза выделяют несколько синдромов.
- Интоксикационный.Его признаки: повышенная утомляемость, выраженная общая слабость, потеря веса, повышение температуры тела, гипергидроз, общая слабость. Лихорадка может быть связана как непосредственно со злокачественным процессом, так и с инфекционными осложнениями.
- Гиперпластический. Лимфобласты с током крови разносятся по организму, накапливаясь в тканях, этот процесс называется лейкемической инфильтрацией. Он проявляется увеличением печени, селезенки, лимфатических узлов, болями в суставах и костях. Лейкемическая инфильтрация оболочек и вещества головного мозга приводит к развитию нейролейкоза. Клинически он проявляется головной болью, тошнотой, иногда рвотой. При осмотре глазного дна отмечают отек дисков зрительных нервов. В некоторых случаях нейролейкоз протекает со стертой клинической картиной или вообще бессимптомно и диагностируется только в ходе лабораторного исследования спинномозговой жидкости. Примерно у 30% мальчиков симптомом лимфобластного лейкоза является образование инфильтратов в яичках. На слизистых оболочках и кожных покровах у больных нередко возникают лейкемиды (инфильтраты багрово-синюшного цвета). В редких случаях гиперпластический синдром проявляется нарушением выделительной функции почек, поражением кишечника и выпотным перикардитом.
- Анемический. Угнетение костномозгового кроветворения сопровождается развитием анемии. У пациентов отмечаются бледность кожных покровов и слизистых оболочек, тахикардия, слабость, головокружение.
- Геморрагический. К развитию этого синдрома приводят тромбозы капиллярных сосудов и тромбоцитопения. На коже появляются множественные петехии и экхимозы. Даже незначительный ушиб сопровождается возникновением обширной подкожной гематомы. Наблюдаются частые носовые, десневые, маточные и желудочно-кишечные кровотечения, кровоизлияния в сетчатку.
- Инфекционный. При лимфобластном лейкозе не происходит полноценной дифференцировки лимфоцитов, в связи с чем они не способны выполнять свои функции, что приводит к значительному снижению иммунитета. Из-за этого пациенты становятся подверженными вирусным, бактериальным и грибковым инфекциям, которые к тому же принимают тяжелое течение и могут приводить к сепсису, инфекционно-токсическому шоку.
Генетические аномалии, способствующие развитию лимфобластного лейкоза, возникают на этапе внутриутробного формирования плода.
Диагностика
Диагностика острого лимфобластного лейкоза осуществляется на основании симптомов заболевания, результатов миелограммы и анализа периферической крови. В общем анализе крови при лимфобластном лейкозе выявляют:
- снижение концентрации гемоглобина (анемия);
- снижение количества тромбоцитов (тромбоцитопения);
- повышенное содержание лейкоцитов (лейкоцитоз), реже наблюдается снижение количества лейкоцитов (лейкопения);
- повышение СОЭ;
- содержание лимфобластов составляет 15–20% от общего числа лейкоцитов;
- снижение количества нейтрофилов (нейтропения).
В миелограмме определяется выраженное угнетение нейтрофильного, эритроидного и тромбоцитарного ростков, преобладание бластных клеток.
В комплексную программу обследования пациентов с лимфобластным лейкозом входят:
- люмбальная пункция с последующим лабораторным исследованием спинномозговой жидкости – для исключения или выявления нейролейкоза;
- рентгенография грудной клетки – с целью обнаружения увеличенных лимфоузлов в средостение;
- УЗИ органов брюшной полости – оценка состояния внутрибрюшных лимфатических узлов и паренхиматозных органов;
- биохимическое исследование крови – для выявления возможных нарушений почек и печени.
Острый лимфобластный лейкоз требует проведения дифференциальной диагностики со следующими патологическими состояниями:
- инфекционным мононуклеозом;
- другими видами лейкоза;
- отравлениями;
- лейкозоподобным синдромом, возникающем на фоне тяжелого течения некоторых инфекционных заболеваний (коклюша, туберкулеза, цитомегаловирусной инфекции, сепсиса).
Лечение лимфобластного лейкоза
Основным методом лечения лимфобластного лейкоза является полихимиотерапия – вид химиотерапии, в которой применяется не один, а несколько цитостатических препаратов.
Взрослые люди заболевают лимфобластным лейкозом в 10 раз реже, чем дети. Показатель заболеваемости возрастает среди пациентов старше 60 лет.
В терапии заболевания выделяют два этапа:
- Интенсивная, или индукционная терапия. Проводится в условиях отделения онкогематолгии на протяжении нескольких месяцев. Противоопухолевые препараты вводят внутривенно. Целью этого этапа является нормализация процессов кроветворения (отсутствие бластов в периферической крови и не более 5% их в костном мозге) и улучшение общего состояния больных.
- Поддерживающая терапия. Проводятся на протяжении нескольких лет в амбулаторных условиях. Противоопухолевые препараты назначают в пероральных формах. Регулярно проводят исследование костного мозга и состава периферической крови пациентов, при необходимости корректируя лечение, например, включая в курс радио- или иммунотерапию.

При низкой эффективности проводимого лечения и повторных обострениях решают вопрос о целесообразности трансплантации костного мозга.
Возможные последствия и осложнения
На фоне острого лимфобластного лейкоза у пациентов происходит значительно снижение гуморального и клеточного иммунитета. В результате у них часто развиваются инфекционно-воспалительные заболевания (тонзиллит, синусит, пиелонефрит, пневмония), которые принимают тяжелое затяжное течение и могут стать причиной сепсиса.
Одной из основных особенностей лимфобластного лейкоза является частая лейкемическая инфильтрация нервных стволов, вещества и оболочек головного мозга, приводящая к развитию нейролейкоза. Без необходимой профилактики это осложнение возникает у каждого второго пациента.
Прогноз
При остром лимфобластном лейкозе у детей прогноз хороший. Современная полихимиотерапия позволяет добиться стойкой ремиссии у 95% пациентов этой возрастной группы. У 70-80% из них длительность ремиссии составляет более 5 лет, таких детей снимают с учета как полностью излечившихся.
При низкой эффективности проводимого лечения и повторных обострениях решают вопрос о целесообразности трансплантации костного мозга.
У взрослых прогноз при лимфобластном лейкозе серьезен, пятилетняя выживаемость не превышает 34–40%.
Профилактика
Специфическая профилактика лимфобластного лейкоза не разработана. Определенную роль в предотвращении заболевания играет здоровый образ жизни (занятия спортом, отказ от вредных привычек, правильное питание, соблюдение режима дня).
Видео с YouTube по теме статьи:
Читайте также: