
Морфология при лимфобластном остром лейкозе
Лейкозы – генерализированные опухоли системы крови с обязательным поражением костного мозга.
Общепризнано, что лейкозы - это полиэтиологические заболевания.
1. Физические факторы: ионизирующее излучение, УФО, вибрация, перепады температур;
2. Химические вещества: воздействие бензола, нитритов, нитратов, пестицидов, лекарств, стероидных гормонов, бутадиона, левомицитина, ароматических аминов, метаболитов триптофана, тирозина, лейцина (более 500 веществ);
3. Вирусы: постоянно находятся в клетках организма в латентном состоянии, под влиянием факторов активируются и происходит мутация клетки. В настоящее время выделен вирус, который вызывает Т- клеточный лейкоз.
4. Иммунологическая недостаточность: врожденная - из-за дефекта вилочковой железы наблюдается склонность к инфекциям, приобретенная - из-за воздействия цитостатиков, облучения;
5. Наследственная предрасположенность: не стабильность хромосом, транслокации хромосом (семейные лейкозы);
6. Неполноценность репаративных процессов в клетках.
Классификация основана не на клинике, а на особенностях морфологического субстрата лейкозов (клеток, составляющих лейкоз).
Острые лейкозы – опухоли, при которых морфологическим субстратом являются бластные клетки, не способные к дифференциации и созреванию в зрелые клетки.
Дифференциация различных видов острых лейкозов основывается на морфологических, иммунологических, цитохимических исследованиях. Более точным является иммунотипирование с помощью моноклональных антител.
Хронические лейкозы – опухоли, при которых лейкозные клетки сохранили способность дифференцироваться и созревать до зрелых форм, морфологическим субстратом являются зрелые и созревающие клетки. Хронические миелопролиферативные заболевания - хронический миелоидный лейкоз (ХМЛ), эритремия, миелофиброз, хронический моноцитарный лейкоз др. Хронические лимфопролиферативные заболевания - хронический лимфоцитарный лейкоз (ХЛЛ), миеломная болезнь и др.
ОСТРЫЕ ЛЕЙКОЗЫ – злокачественные опухоли, происходящие из стволовых или близких к ней клеток - предшественниц (1-4 классов). Морфологическим субстратом являются бластные клетки, утратившие способность к дифференцировке и созреванию или сохранившие способность к извращенной псевдодифференциации до клеток, напоминающих миелобласты, лимфобласты.
Лейкоциты – у 50 – 70% лейкоцитоз от 10 х 10 9 /л до 400 х 10 9 /л за счет бластов;
– у 30 – 50% лейкопения или норма.
Лейкоцитарная формула – преобладают бластные клетки, иногда до 90% и небольшой процент зрелых клеток. Бласты обычные, типичные или выраженная атипия: крупные или мелкие с дегенеративными изменениями, очень хрупкие, ломкие.
Лейкемическое зияние = лейкемический аборт = провал – это состояние в крови или костном мозге, когда при подсчете лейкограммы или миелограммы встречается большое количество бластов и немного зрелых клеток, созревающих клеток нет. Признак острого лейкоза.
Анемия. Количество эритроцитов и гемоглобина снижено, нормохромия, нормоциты, ретикулоцитопения.
Тромбоциты значительно снижены.
Биохимия: Повышение концентрации мочевой кислоты в сыворотке и моче как критерий интоксикации. Диспротеинемия.
Пункция костного мозга: В 95 % случаев наблюдается тотальная бластная гиперплазия костного мозга, более 30 % бластов, лейкемическое зияние выражено, дегенеративные изменения в лейкоцитах и других клетках.
МОРФОЛОГИЯ ЛЕЙКОЗНЫХ КЛЕТОК:
1. Размеры: увеличение (в 2 –3 раза) или уменьшение, анизоцитоз лейкоцитов.
2. Ядро: ядерно-цитоплазматическое соотношение увеличивается в пользу ядра. Контуры ядра деформированы, количество хроматина повышено, распределено неравномерно, вакуолизация, сегментация, стадии митоза.
3. Нуклеолы: число их увеличено (8 и более) размеры ядрышек составляют 1/3 – 1/2 диаметра ядра, если более 1/3 – признак злокачественности клетки.
4. Цитоплазма: повышается ее базофильность, вакуолизация, иногда вакуоли содержат зернышки. При остром миелобластном лейкозе – азурофильная зернистость и образования в форме палочек, напоминающие кристаллы – тельца Ауэра.
Хронические миелопролиферативные заболевания: хронический миелоидный лейкоз (ХМЛ), морфологический субстрат, особенности лейкозных клеток, лабораторная диагностика, картина крови.
Хронический миелоидный лейкоз (ХМЛ) – опухоль системы крови, возникающая из мутировавшей стволовой клетки или близкой к ней клетки-предшественницы. Клетки возникшего опухолевого клона сохраняют способность дифференцироваться и созревать до зрелых форм. Основной морфологический субстрат лейкоза – созревающие и зрелые гранулоциты.
По морфологическим характеристикам лейкемические гранулоциты вначале существенно не отличаются от нормальных, но принципиально отличаются в функциональном отношении. В их цитоплазме снижена активность ферментов (щелочной фосфатазы, миелопероксидазы и др.), характерна более мелкая зернистость, нарушена фагоцитарная активность клеток.
РАЗВЕРНУТАЯ СТАДИЯ. ЛАБОРАТОРНАЯ ДИАГНОСТИКА:
Диагноз ХМЛ устанавливают по данным анализа крови.
Концентрация гемоглобина и количество эритроцитов, как правило, в пределах нормы, однако у части больных уже могут быть признаки анемии или эритроцитоза.
Наиболее характерным для ХМЛ является лейкоцитоз 20,0 - 30,0 х 10 9 /л и более (до 600 х 10 9 /л), главным образом за счет нейтрофилов разной степени зрелости.
Наблюдается сдвиг влево до единичных промиелоцитов, у некоторых больных – до бластных клеток. Количество миелоцитов и метамиелоцитов (юных), как правило, значительное – 5 % и более. Количество палочкоядерных нейтрофилов небольшое.
Характерный гематологический признак ХМЛ – увеличение количества эозинофилов и базофилов (эозинофильно-базофильная ассоциация).
В некоторых случаях увеличивается только число эозинофилов или базофилов. Большое количество базофилов – плохой прогностический признак.
Морфология гранулоцитов на этапе диагностики ХМЛ существенно не изменена. Самое частое нарушение (уменьшение количества и размера гранул в цитоплазме нейтрофилов (гипо- и агрануляция нейтрофилов). При подсчете лейкоцитарной формулы эту особенность нейтрофилов следует учитывать, так как миелоциты, метамиелоциты можно принять за лимфоциты или моноциты, тем более что, цитоплазма нейтрофилов может отставать по степени зрелости от ядра (диссоциация в созревании ядра и цитоплазмы) и окрашиваться в сиреневый или голубовато-сиреневый цвет.
СОЭ – чаще в пределах нормы или соответствует клиническому состоянию больного (инфекция, анемия или др.).
Количество тромбоцитов соответствует норме, у некоторых больных может наблюдаться тромбоцитоз или тромбоцитопения. Отклонение от нормы количества тромбоцитов неблагоприятно в прогностическом отношении.
Пункция костного мозга не обязательна. Выявляют гиперплазию гранулоцитарного ряда, коэффициент лейко/эритро составляет 20:1, 30:1. Костный мозг повторяет периферическую кровь.
КРИТЕРИИ ДИАГНОСТИКИ ХМЛ:
1. клиническая картина: относительно хорошее самочувствие, умеренное увеличение селезенки, в некоторых случаях и печени;
2. лейкоцитоз с тенденцией к росту за счет гранулоцитов;
3. сдвиг нейтрофилов влево до промиелоцитов или бластов;
4. отсутствие грубых дегенеративных изменений нейтрофилов (токсигенной зернистости и др.);
5. повышенное количество эозинофилов и (или) базофилов;
6. в костном мозге выявляется гиперплазия гранулоцитарного ростка;
7. обнаружение Рh΄ – хромосомы и онкогена BCR/ABL.
В сыворотке крови больных ХМЛ выявляют повышенный уровень витамина В12, гистамина, мочевой кислоты (часто пропорционально степени интоксикации).
Относительно доброкачественная моноклоновая опухоль превращается в злокачественную поликлоновую. Появляющиеся новые клоны злокачественных клеток уже не могут дифференцироваться и созревать до зрелых гранулоцитов. В крови, обнаруживается большое количество бластных клеток. Такое явление называется бластным кризом. Лейкоз вступает в терминальную стадию.
ТЕРМИНАЛЬНАЯ СТАДИЯ. ЛАБОРАТОРНАЯ ДИАГНОСТИКА:
При исследовании крови выявляют анемию нормо– или гиперхромного характера.
Количество лейкоцитов различно – от умеренного до чрезвычайно высокого от 700,0 х 10 9 /л до 1000,0 х 10 9 /л.
В лейкоцитарной формуле 10 – 99% бластов, чаще десятки процентов. Характерен полиморфизм бластов, их атипия.
Тромбоцитоз до 1500 х 10 9 /л или тромбоцитопения.
Миелемия – состояние, когда кровь повторяет косный мозг.
Острые лейкемии часто имеют много общих цитоплазмати-ческих и мембранных характеристик. Это позволяет рассматривать разные типы острых лейкемий практически как специфическую стадию созревания миелоидных и лимфоидных клеток. Упрощенная схема представлена на рисунке ниже.
Диагноз ставится на основании анализа крови и рентгена костного мозга. В типичных случаях лейкемические нарушения в крови и костном мозге приводят к образованию однообразных бластных клеток. В случаях ОЛЛ почти всегда отмечают их появление, но у взрослых при ОМЛ лейкемический процесс может быть настолько неуловим, что обнаруживают лишь незначительное количество бластов в крови.
Для постановки достоверного диагноза необходимо, чтобы в костном мозге присутствовало до 30% бластных клеток. Если в костном мозге присутствует менее 20% бластов, обычно ставят диагноз миелодисплазия.
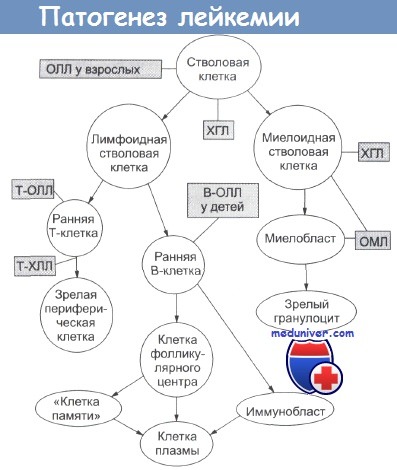
Созревание кроветворных и лимфоидных клеток при различных типах лейкемии.
Острые лейкемии подразделяют на морфологические типы, используя схему, предложенную группой французских, американских и английских исследователей. Основные принципы классификации даны в таблице ниже.
В случае ОМЛ миелобласты могут содержать палочки Ауэра, розоватые кристаллические цитоплазматические включения, которые возможно являются отклонением от нормы нормальных цитоплазматических гранул, обнаруживающихся в клетках-предшественниках гранулоцитов в норме. Эти гранулы часто видны в цитоплазме при ОМЛ и практически обычны, и нередко их много в случае МЗ (гипергранулярная промиелоцитарная лейкемия).
В случае моноцитарной лейкемии (М5) обнаруживают много бласт с избыточной цитоплазмой, при эритролейкемии (М6) в костном мозге выявляются эритробласты и миелобласты.

Морфологическая классификация острых лейкемий. Доля подтипов острых лейкемий миелоидного и моноцитарного происхождения (%).
Наиболее распространенная форма ОЛЛ (L1) характеризуется наличием в бластных клетках скудной цитоплазмы и немногочисленных ядрышек. В менее распространенном случае лейкемии L2 бласты крупнее с более крупным ядрышком. Эти формы заболевания, которые встречаются чаще у взрослых, легко спутать с ОМЛ, так что в некоторых случаях говорят о недифференцированной лейкемии. При лейкемии L3 обнаруживают большие гомогенные клетки с видимыми ядрышками и базофильной цитоплазмой, похожие на клетки, выявляемые при лимфоме Беркитта.
В сомнительных случаях определить тип лейкемии помогает цитохимическое окрашивание. Наиболее часто используемые типы окрашивания представлены в таблице ниже. Эти красители могут помочь в установке диагноза в сомнительных случаях. В редких случаях недифференцированной острой лейкемии цитохимическое окрашивание не помогает. Маркеры, при ОЛЛ в особенности, помогают дифференцировать общий случай ОЛЛ от Т-клеточной ОЛЛ и лимфомы Беркитта.

Примечание. TCR — Т-клеточный рецептор;
Tdt — ДНК-полимераза, определенная методами иммунофлуоресценции или биохимически.
Нормальная последовательность созревания лимфоидных и миелоидных клеток сопровождается биосинтезом цитоплазматических белков и белков поверхности клетки, которые можно выявить антителами. При лейкемии используют экспрессию антигенов для фенотипирования опухолей разных стадий созревания.
Некоторые из антигенов имеют незначительную прогностическую и терапевтическую ценность, но они помогают понять процесс патогенеза при лейкемии. Краткая информация с сокращениями представлена в таблице ниже.

Примечание. Могут применяться и некоторые другие маркеры. Здесь представлена широко используемая панель маркеров.
В некоторых случаях маркеры используют по морфологическим и клиническим показаниям.
су - цитоплазматический; SIg - иммуноглобулин клеточной поверхности; tdt - терминальная дезоксирибонуклеотидтрансфераза.
ОСТРЫЕ ЛЕЙКОЗЫ
Вариантами острого лейкоза являются миелобластный, лимфобластный, монобластный, промиелоцитарный, плазмобластный,мегакариобластный, эритромиелоз, недифференцированный и др.
При острых миелобластных лейкозах число лейкоцитов крови может достигать (100-150) х 10 9 /л и выше; часто оно держится на уровне (20-30) х 10 9 /л и ниже. В мазке крови наряду с незначительным количеством зрелых сегментированных нейтрофилов нередко все поле зрения усеяно миелобластами.
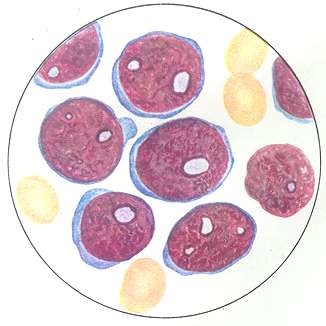
Большое содержание бластных клеток в периферической крови
У детей чаще всего (80 % случаев) встречается острый лимфобластный лейкоз. Другие формы острых лейкозов наблюдаются значительно реже. Хроническим миелолейкозом дети болеют редко (5% лейкозов).
При остром лимфобластном лейкозе в крови преобладают лимфобласты (рис.50).

Картина крови при остром лимфобластном лейкозе: в периферической крови присутствует большое количество бластных клеток.
ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ
Среди хронических лейкозов выделяют миелолейкоз, лимфолейкоз, моноцитарный и миеломную болезнь (плазмоцитома) и др.
При хроническом миелолейкозе наряду с резким повышениемобщего количества лейкоцитов(до 600 х 10 9 /л) наблюдается появление в периферической крови огромного количества гранулоцитов на ранних стадиях развития: миелобластов, промиелоцитов, миелоцитов, метамиелоцитов.

Картина крови при хроническом миелолейкозе: 1 – нейтрофильный промиелоцит;
2 – миелоциты;
3 – метамиелоцит;
4 – палочкоядерный лейкоцит;
5 – сегментоядерный лейкоцит
Процентное содержание зрелых и созревающих нейтрофилов в лейкоцитарной формуле становится преобладающим. При хронических миелолейкозах в крови имеет место также повышенное количество эозинофильных и базофильных миелоцитов (эозинофильно-базофильная ассоциация).
В редких случаях миелолейкоза лейкемическая картина крови может наблюдаться при нормальном и даже пониженном общем количестве лейкоцитов в крови. В других случаях картина крови может быть нормальной, а диагноз лейкоза ставится лишь при исследовании пунктата костного мозга (алейкемический миелолейкоз).

Бластный криз при хроническом миелолейкозе
При хроническом лимфолейкозепреобладают зрелые лимфоциты, присутствуют единичные лимфобласты, тени Боткина-Гумпрехта, представляющие собой разрушенные при приготовлении мазка лимфоциты:

Картина крови при хроническом лимфолейкозе: * – тени Боткина-Гумпрехта
Дата добавления: 2018-08-06 ; просмотров: 595 ;
Что такое острый лимфобластный лейкоз?
Острый лимфобластный лейкоз (или острый лимфолейкоз, сокр. ОЛЛ) — это опасное для жизни заболевание, при котором клетки, в нормальных условиях развивающиеся в лимфоциты, становятся злокачественными и быстро замещают нормальные клетки в костном мозге.
- В связи с недостатком нормальных клеток крови у больных могут появляться такие симптомы, как повышение температуры, слабость и бледность.
- Как правило, в таких случаях выполняются анализы крови и исследование костного мозга.
- Проводится химиотерапия, которая часто оказывается эффективной.
Острый лимфолейкоз (ОЛЛ) встречается у больных любого возраста, но является самым распространенным типом онкологических заболеваний у детей и составляет 75% всех случаев лейкоза у детей до 15 лет. ОЛЛ наиболее часто поражает детей младшего возраста (от 2 до 5 лет). Среди людей среднего возраста это заболевание встречается немного чаще, чем у больных старше 45 лет.
При ОЛЛ очень незрелые лейкозные клетки скапливаются в костном мозге, разрушая и замещая клетки, которые производят нормальные клетки крови. Лейкозные клетки переносятся с кровотоком в печень, селезенку, лимфатические узлы, головной мозг и яички, где могут продолжать расти и делиться. При этом клетки ОЛЛ могут скапливаться в любой части организма. Они могут проникать в оболочки, покрывающие головной и спинной мозг (лейкозный менингит), и приводить к анемии, печеночной и почечной недостаточности и повреждению других органов.
Симптомы острого лимфобластного лейкоза

Ранние симптомы ОЛЛ возникают в результате неспособности костного мозга вырабатывать достаточное количество нормальных клеток крови.
- Повышение температуры и чрезмерное потоотделение могут указывать на наличие инфекции. Высокий риск инфекции связан со слишком малым количеством нормальных лейкоцитов.
- Слабость, утомляемость и бледность, свидетельствующие об анемии, могут появляться из-за недостаточного количества эритроцитов. У некоторых больных может наблюдаться затрудненное дыхание, учащенное сердцебиение и боль в груди.
- Быстро появляющиеся кровоподтеки и кровотечения, иногда в форме носовых кровотечений или кровотечений из десен, возникают из-за слишком малого количества тромбоцитов. В некоторых случаях может возникать кровоизлияние в головной мозг или внутрибрюшное кровотечение.
При проникновении лейкозных клеток в другие органы возникают соответствующие симптомы.
- Лейкозные клетки в головном мозге могут вызывать головные боли, рвоту, инсульт и нарушения зрения, равновесия, слуха и лицевых мышц.
- Лейкозные клетки в костном мозге могут приводить к болям в костях и суставах.
- Если лейкозные клетки вызывают увеличение печени и селезенки, может появляться ощущение переполнения желудка и в некоторых случаях боль.
Причины острого лимфобластного лейкоза
Основная причина ОЛЛ остается неизвестной, но существуют факторы риска, которые могут быть экологическими или вторичными по отношению к наследственным и/или приобретенным предрасполагающим условиям. Факторами риска окружающей среды являются прошлое облучение ионизирующим излучением, химическими веществами (бензол, гербициды и пестициды) и химиотерапевтическими агентами.
К наследственным предрасполагающим состояниям относятся синдром Дауна, наследственные расстройства, характеризующиеся дефектом процессов репарации ДНК и регуляции клеточного цикла (анемия Фанкони, синдром Блума и атаксия-телеангиэктазия), наследственные расстройства, характеризующиеся изменением передачи сигнала в процессах пролиферация клеток и апоптоз (синдром Костмана, синдром Швахмана–Даймонда, анемия Даймонда–Блекфена и нейрофиброматоз типа I) и синдром Ли-Фраумени.
Существуют также приобретенные предрасполагающие состояния, такие как апластическая анемия, пароксизмальная ночная гемоглобинурия и миелодиспластический синдром.
Диагностика
Первые признаки острого лимфобластного лейкоза можно обнаружить при помощи анализов крови, таких, как общий анализ крови. Общее количество лейкоцитов может быть сниженным, нормальным или повышенным, но количество эритроцитов и тромбоцитов почти всегда оказывается сниженным. Кроме того, в крови обнаруживаются очень незрелые лейкоциты (бласты).
Чтобы подтвердить диагноз и отличить ОЛЛ от других типов лейкозов, практически во всех случаях проводится исследование костного мозга. Бласты анализируют на наличие хромосомных аномалий, что помогает врачам определить точный тип лейкоза и подобрать подходящие препараты для лечения.
Анализы крови и мочи назначаются для выявления других отклонений, включая электролитные нарушения.
Также могут потребоваться визуализирующие исследования. При выявлении симптомов, позволяющих заподозрить наличие лейкозных клеток в головном мозге, проводится компьютерная томография (КТ) или магнитно-резонансная томография (МРТ). Для выявления лейкозных клеток в области вокруг легких может выполняться КТ органов грудной клетки. При увеличении внутренних органов могут проводиться КТ, МРТ или ультразвуковое исследование брюшной полости. Перед началом химиотерапии может быть выполнена эхокардиография (ультразвуковое исследование сердца), поскольку иногда химиотерапия оказывает отрицательное воздействие на сердце.
Лечение острого лимфобластного лейкоза
Лечение ОЛЛ включает:
- химиотерапию;
- другие препараты, такие как иммунотерапия и/или таргетная терапия;
- в редких случаях трансплантация стволовых клеток или лучевая терапия.
Химиотерапия является высокоэффективной и состоит из следующих фаз:
- индукция;
- лечение головного мозга;
- консолидация и интенсификация;
- поддерживающая терапия.
Индукционная химиотерапия — это первая фаза лечения. Задача индукционной терапии состоит в достижении состояния ремиссии посредством уничтожения лейкозных клеток, что восстанавливает способность нормальных клеток развиваться в костном мозге. В некоторых случаях требуется пребывание в больнице в течение нескольких дней или недель (это зависит от того, насколько быстро восстанавливается костный мозг).
Применяется одна из нескольких комбинаций лекарственных препаратов, дозы которых вводятся повторно в течение нескольких дней или недель. Выбор конкретной комбинации зависит от результатов диагностических анализов. Одна из комбинаций состоит из преднизона (кортикостероида), принимающегося внутрь, и еженедельных доз винкристина (химиотерапевтического препарата), назначаемого вместе с препаратом антрациклина (обычно даунорубицином), аспарагиназой и иногда циклофосфамидом, для внутривенного введения. У некоторых пациентов с острым лимфолейкозом могут использоваться новые препараты, такие как иммунотерапия (лечение, которое использует собственную иммунную систему человека для уничтожения опухолевых клеток) и таргетная терапия (препараты, которые атакуют внутренние биологические механизмы опухолевых клеток).
Лечение головного мозга обычно начинается во время индукции и может продолжаться на всех этапах лечения. Поскольку ОЛЛ часто распространяется и на головной мозг, эта фаза также направлена на лечение лейкоза, уже распространившегося в головной мозг, либо на профилактику распространения лейкозных клеток в головной мозг. Для воздействия на лейкозные клетки в слоях ткани, покрывающих головной и спинной мозг (мозговых оболочках), применяются лекарственные препараты, такие, как метотрексат, цитарабин, кортикостероиды или их комбинации, которые обычно вводятся прямо в спинномозговую жидкость, либо высокие дозы этих препаратов могут вводиться внутривенно. Такая химиотерапия может проводиться в сочетании с лучевой терапией головного мозга.
В фазе консолидации и интенсификации продолжается лечение заболевания костного мозга. Дополнительные химиотерапевтические препараты или те же препараты, что и во время фазы индукции, могут применяться несколько раз за период, который продолжается в течение нескольких недель. Некоторым больным с высоким риском рецидива в связи с определенными хромосомными изменениями в лейкозных клетках назначают пересадку стволовых клеток после достижения ремиссии.
Дальнейшая поддерживающая химиотерапия, которая обычно заключается в приеме меньшего количества препаратов (в некоторых случаях в меньших дозах), продолжается, как правило, в течение 2–3 лет.
Пожилые люди с ОЛЛ могут быть не способны перенести интенсивную схему лечения, используемую у молодых людей. У таких больных может быть использован более щадящий вариант лечения с применением только режимов индукционной терапии (без последующей консолидации, интенсификации или поддерживающей терапии). Иногда у некоторых пожилых людей может назначаться иммунотерапия или более щадящая форма трансплантации стволовых клеток.
Во время всех вышеуказанных фаз для лечения анемии и предотвращения кровотечений может потребоваться переливание крови и тромбоцитов, а для лечения инфекций — прием противомикробных препаратов. Чтобы помочь избавить организм от вредных веществ (таких как мочевая кислота), которые образуются при разрушении лейкозных клеток, могут проводиться внутривенные вливания жидкостей и лечение препаратами аллопуринол либо расбуриказа.
Лейкозные клетки могут начать появляться снова (такое состояние называют рецидивом). Часто они образуются в крови, костном мозге, головном мозге или яичках. Раннее повторное появление таких клеток в костном мозге является особенно серьезным. Химиотерапия проводится еще раз, и, хотя многим больным помогает такое повторное лечение, существует большая вероятность повторного рецидива заболевания, особенно у детей первого года жизни и взрослых. Если лейкозные клетки повторно появляются в головном мозге, химиотерапевтические препараты 1 или 2 раза в неделю вводятся в спинномозговую жидкость. Если лейкозные клетки повторно появляются в яичках, то наряду с химиотерапией проводится лучевая терапия на область яичек.
У некоторых пациентов с рецидивирующим ОЛЛ используются новые перспективные методы лечения с использованием моноклональных антител (белков, которые специфически связываются с лейкозными клетками, маркируя их для уничтожения). Еще более новая терапия, которую можно применять у некоторых пациентов с рецидивом острого лимфобластного лейкоза, называется Т-клеточная терапия с химерным антигенным рецептором (CAR-T). Эта терапия предусматривает модификацию определенного вида лимфоцитов (Т-лимфоцитов, также называемых Т-клетками) от больного лейкозом таким образом, чтобы эти новые Т-лимфоциты лучше распознавали и атаковали лейкозные клетки.
После рецидива у больных, неспособных перенести пересадку стволовых клеток, дополнительная терапия часто оказывается плохо переносимой и неэффективной и обычно приводит к серьезному ухудшению самочувствия. Тем не менее, могут случаться ремиссии. В отношении пациентов, которым не помогает лечение, должен рассматриваться вариант ухода за неизлечимо больными людьми.
Прогноз жизни
До появления лечения большинство больных с острым лимфолейкозом умирали в течение нескольких месяцев с момента постановки диагноза. Теперь ОЛЛ удается излечить примерно у 80% детей и у 30–40% взрослых. У большинства больных первый курс химиотерапии позволяет взять заболевание под контроль (полная ремиссия). Лучшие прогнозы на излечение имеются у детей в возрасте 3-9 лет. Прогнозы для детей первого года жизни и пожилых больных менее благоприятны. Количество лейкоцитов на момент установления диагноза, наличие или отсутствие распространения лейкоза в головной мозг и хромосомные аномалии в лейкозных клетках также влияют на результат лечения.
Острые миелоидные лейкозы
Острые миелоидные лейкозы (ОМЛ) представляют гетерогенную группу клональных заболеваний, при которых неопластическая трансформация наблюдается в мультилинейной стволовой клетке или в стволовой клетке с линейной дифференцировкой.
Поскольку мультипотентная стволовая клетка дает начало гранулоцитам, моноцитам, эритроцитам и мегакариоцитам, при ОМЛ возможно поражение всех или отдельных клеточных линий.
В целом неопластический клон прекращает дифференцировку на стадии бласта, что ведет к прогрессирующему накоплению бластов в костном мозге (КМ) с последующим их выходом в периферическую кровь (Рис. 1, 2).

Рис. 1. Схема кроветворения

Рис. 2. Мезенгиогенез. Мезенхимальная стволовая клетка (МСК)
ОМЛ составляют около 20% от всех лейкозов и диагностируются в любом возрасте, однако частота их возникновения увеличивается с возрастом, медиана возраста составляет 60-65 лет.
Заболеваемость составляет 2-3 человека на 100000 населения в год. Этиология острого миелоидного лейкоза неизвестна, однако причинными факторами могут быть: алкилирующие препараты, некоторые химические вещества, пестициды, рентгенконтрастные вещества, красители, миелотоксические агенты, ионизирующее излучение.
Дозы, превышающие 100 рад, оказывают дозо-зависимый эффект в отношении возникновения ОМЛ. Генетическая аномалия - трисомия Хр21 (синдром Дауна) вызывает повышенную частоту развития ОМЛ, хотя в большинстве случаев при этом синдроме развивается острый лимфобластный лейкоз (ОЛЛ).
Повышен риск развития ОМЛ при синдроме Блума, анемии Фанкони, атаксии-телеангиэктазии, нейрофиброматозе Реклингаузена и т.п. Гематологические нарушения - апластическая анемия, хронический миелопролиферативный синдром, миелодиспластические состояния, пароксизмальная ночная гемоглобинурия повышают риск развития острого миелоидного лейкоза.
Клинические проявления неспецифичны. Жалобы на общую слабость, недомогание, головокружение могут задолго предшествовать диагнозу. Нередко отмечается бледность кожных покровов, повышение температуры тела без наличия инфекции, довольно часто отмечается геморрагический синдром по петехиально-пятнистому типу, иногда кровотечения.
Оссалгии отмечаются у 20% больных. Гепатоспленомегалия не является диагностическим признаком ОМЛ, но отмечается у 50% больных. Лейкемиды кожи и инфильтрация десен обычно характерны для миеломоно- и монобластного варианта. Исходное поражение ЦНС встречается редко - при гиперлейкоцитозе и/или монобластном варианте ОМЛ.
Цитохимическими маркерами бластов гранулоцитарного ряда являются липиды, миелопероксидаза (МПО), ASD-хлорацетатэстераза (ASD-ХАЭ). Определение последнего фермента имеет небольшую диагностическую ценность вследствие невысокой ее активности. В диагностике М5а и М5в вариантов ОМЛ главную роль играет исследование неспецифической эстеразы (НЭ), подавляемой фторидом натрия (NaF), антилизоцима.
Применение иммунофенотипирования для диагностики острого миелоидного лейкоза позволяет определить линейность и/или этап дифференцировки бластов, начиная с уровня стволовых клеток-предшественников. Изучение кариотипа бластных клеток позволило выявить закономерные изменения, характерные для отдельных вариантов острых лейкозов.
При цитогенетическом исследовании у 70-80% больных ОМЛ можно выявить неслучайные приобретенные хромосомные аберрации. Некоторые из них, при которых образуется химерный сливной белок, могут служить маркером наличия опухолевого клона. Данные цитогенетического исследования используются для уточнения отдельных вариантов ОМЛ, определения прогноза заболевания, а также для контроля качества и эффективности проводимой терапии.
Таблица 1. Характеристика бластов при ОМЛ (М.А.Френкель, 2001)

Согласно FAB-классификации, выделяют следующие варианты острого миелоидного лейкоза.
Острые миелобластные лейкозы (М0, М1, М2). Этот термин объединяет три подтипа заболевания, которые отличаются друг от друга по степени дифференцировки лейкемических клеток -миелобластов.
М0 составляет около 3-5% ОМЛ. Диагноз может быть установлен только при выполнении иммунофенотипирования. Бласты экспрессируют CD13, CD31. С помощью моноклональных антител (антимиелопероксидазы) - анти-МПО в бластах можно обнаружить фермент миелопероксидазу.
М1 составляет около 15% всех ОМЛ. Морфологически бласты имеют круглое или овальное ядро с рыхлым хроматином, несколько нуклеол и небольшой ободок серо-голубой цитоплазмы без гранул. Тельца Ауэра (первичные лизосомы) выявляются в 10% случаев. При этом варианте определяется минимальная степень миелоидной дифференцировки.
Иммунофенотипическими маркерами является выявление антигенов CD13, 33, 34, HLA-DR. В единичных клетках выявляется транслокация хромосом (Хр) (t 9; 22) и инверсия Хр3. МПО и/или липиды содержатся более чем в 3% бластов.
М2 составляет около 25-32% всех ОМЛ. Более 10% бластов содержит зернистость в цитоплазме в виде нежных темно-красных гранул. Бласты содержат МПО, липиды, гранулоцитарную эстеразу (ГЭ) в большинстве клеток, ШИК-вещество в диффузной форме; бласты экспрессируют антигены CD11, 13, 15, 33, анти-МПО. Примерно в 20-40% случаев выявляется t (8; 21); при этом отмечаются спленомегалия, хлоромы, эозинофилия в периферической крови.
При данной транслокации на длинном плече Хр8 образуется химерный ген AML1-ETO, продуктом деятельности которого является патологический белок CBFa-ETO. При связывании его с ДНК происходит ингибирование транскрипции и, соответственно, нарушаются механизмы дифференцировки миелоидных клеток.
М2 базофильно-клеточный представлен в единичных случаях. Бласты содержат грубую базофильную зернистость, не содержат миелопероксидаза, липиды и ГЭ. Цитогенетически в этих случаях может выявляться t (6; 9). В сыворотке крови повышено содержание гепарина и серотонина. При этой форме острого лейкоза прогноз крайне неблагоприятный.
М3 составляет около 10% случаев острого миелоидного лейкоза. Ядра бластов имеют характерную лопастную форму, цитоплазма содержит обильную азурофильную зернистость и палочки Ауэра. В зависимости от величины гранул выделяют макрогранулярный и микрогранулярный (M3v) варианты данного заболевания. Бласты содержат большое количество МПО, липидов, ГЭ, умеренное количество НЭ, неподавляемой NaF; ШИК-вещество в диффузной форме.
Поскольку в гранулах и тельцах Ауэра содержатся сульфатированные мукополисахариды, обладающие активностью типа тканевого тромбопластина, в большинстве случаев развивается ДВС-синдром (диссеминированное внутрисосудистое свертывание). Бласты экспрессируют антигены CD 11, 13, 15, 33, анти-МПО. В 1977 г. J.D.Rowley и соавторы установили, что в 95% случаев острого промиелоцитарного лейкоза (ОПЛ) обнаруживается t (15; 17).
Вследствие этого PML-ген (ген промиелоцитарного лейкоза), расположенный на Хр15, переносится на длинное плечо Хр17 в область, где находится ген а-рецептора ретиноевой кислоты (RARA). В норме этот ген участвует в регуляции дифференцировки миелоидных клеток. Ген PML является регулятором роста и играет роль в созревании и активации различных клеток.
Продуктом химерного гена PML-RARA является патологический белок, который накапливается в цитоплазме и ядре миелоидных клеток, что приводит к блоку дифференцировки клеток на уровне промиелоцитов. Кроме того, этот белок блокирует механизмы апоптоза, что поддерживает жизнеспособность опухолевых клеток.
В М4 варианте ОМЛ выделяют биклональный (1), бифенотипический (2) и миело-монобластный эозинофильный М4ЕО (3) подварианты. Биклональный вариант составляет около 17% всех случаев, при нем бласты представлены двумя типами клеток -миелобластами и монобластами, и экспрессируют антигены анти-МПО, анти-лизоцим, CD 13, 14, 15, 33. Цитогенетические исследования могут выявить t (8; 21).
Бифенотипический вариант крайне редок. Морфологически бласты характеризуются как миелобласты при М2 варианте, однако во всех бластах одновременно содержится МПО, ГЭ и НЭ, подавляемая NaF.
М5а составляет около 10% острого миелоидного лейкоза. Бласты не имеют специфических морфологических признаков, содержат НЭ, подавляемую фторидом NaF. Отдельные бласты содержат небольшое количество миелопероксидазы и/или липидов. Бласты экспрессируют антигены CD 13, 14, 15, 33, антилизоцим. Иногда (часто после проведенной полихимиотерапии) выявляется аномалия Хр11.
М5в встречается редко. Морфологически бласты имеют моноцитоидную форму ядер, содержат значительное количество НЭ, подавляемой NaF, небольшое количество МПО или липидов. Бласты экспрессируют антигены CD 13, 14, 15, 33, антилизоцим. Как и в предыдущем варианте, может выявляться аномалия Хр11.
Мб (1) - острый эритробластный лейкоз. Характеризуется наличием расширенного патологического красного ростка. Бласты имеют морфологические и цитохимические признаки миелобластов варианта М1. Клетки красного ряда имеют мегалобластоидные изменения с признаками морфологической и цитохимической дисплазии, содержат тельца Жолли, усилена сидерофилия.
Мб (2) - эритромиелоз. В костном мозге преобладает бластная популяция, которая состоит из миелобластов и эритробластов. Бласты не имеют специфических морфоцитохимических признаков, экспрессируют гликофорин А - эритроидный антиген. Цитогенетически можно выявить множественные хромосомные перестройки. Есть мнение, что изменения со стороны клеток красного ряда представляют реакцию на уже имеющийся ОМЛ. Цитогенетические исследования показывают наличие аномалий Хр5,7.
М7 встречается крайне редко. Мегакариобласты в большом количестве в костном мозге с их выходом в периферическую кровь. Цитохимически в бластах находят большое содержание а-нафтил-ацетатэстеразы, не ингибируемой NaF, и некоторое количество а-бутиратэстеразы, выявляется положительная ШИК-реакция. Бласты экспрессируют тромбоцитарные антигены CD 41 и/или CD 61.
Острые лимфобластные лейкозы
Острые лимфобластные лейкозы представляют гетерогенную группу клональных заболеваний лимфопоэтической ткани, характеризующуюся накоплением лимфобластов в КМ. Прогрессирующая инфильтрация опухолевыми лимфобластами костного мозга подавляет продукцию нормальных гемопоэтических клеток. ОЛЛ составляют 10% от всех лейкозов, причем 60% от этого количества наблюдается у детей.
В Европе ОЛЛ является наиболее частым опухолевым заболеванием детского возраста. Пик заболеваемости приходится на возраст 3-7 лет, второй, не очень выраженный пик, отмечается в возрасте 50-60 лет. Заболеваемость острым лимфобластным лейкозом ниже на Среднем Востоке и в Азии. В США частота заболеваемости белого населения в 2 раза выше, чем негроидного.
Подтип В-клеточного ОЛЛ составляет 80% всех случаев ОЛЛ, почти во всех остальных случаях диагностируется Т-ОЛЛ. Крайне редко определяется нуль-клеточный вариант.
Этиология ОЛЛ неизвестна. Не выявлено связи с миелотоксическими агентами, химикатами, ретровирусами, хотя внедрение ретровируса в геном при лечении агаммаглобулинемии Бруттона в некоторых случаях приводило к развитию острого лимфобластного лейкоза.
Умеренное повышение заболеваемости ОЛЛ отмечено под воздействием высоких доз ионизирующего излучения и при некоторых иммунодефицитных состояниях. Хотя вирус Эпштейн-Барра инкорпорируется в геном клеток при африканском варианте лимфомы Беркитта, вирусный геном отсутствует при лимфобластном L3 варианте ОЛЛ. Заболеваемость ОЛЛ в 15-20 раз выше при синдроме Дауна.
В патогенезе ОЛЛ основным является прогрессирующее накопление лимфобластов в КМ, что снижает потенциал дифференцировки и созревания. Для популяции лейкемических лимфобластов характерна остановка дифференцировки трансформированных лимфопоэтических стволовых клеток на специфической стадии, что снижает также уровень апоптоза.
Лейкемические лимфобласты имеют удлиненное время генерации (3 суток) в сравнении с нормальными лимфоидными предшественниками (1 сутки). Поскольку лимфобласты не созревают до более дифференцированной стадии, они накапливаются в костном мозге. Клинически это проявляется анемией, гранулоцитопенией и тромбоцитопенией.
Клиника ОЛЛ обычно представлена неспецифическими симптомами, хотя дебют может быть острым. Наиболее часто отмечаются слабость, сонливость, оссалгии, артралгии, не связанное с инфекцией повышение температуры тела. Иногда единственной жалобой являются оссалгии при отсутствии лимфоаденопатии, органомегалии и изменений в анализах периферической крови.
При вовлечении в процесс ЦНС отмечаются головная боль, тошнота, рвота. Бактериальная инфекция встречается при выраженной нейтропении. При физикальном обследовании может отмечаться бледность кожных покровов, геморрагический синдром по петехиально-пятнистому типу, лимфоаденопатия (чаще в области шеи), гепатоспленомегалия. Крайне редко бывают лейке -миды кожи.
В анализе крови в 60% случаев отмечается сублейкоцитоз, в 10% - гиперлейкоцитоз свыше 100,0х10 9 /л, тромбоцитопения менее 50,0х10 9 /л - у 60% больных в момент диагностики. При гиперлейкоцитозе почти всегда отмечается лимфоаденопатия, гепато- и спленомегалия. Анемия является непостоянным признаком.
В пунктате КМ отмечается гиперклеточность с наличием более 20% бластов. Для установления диагноза необходимо комплексное (морфологическое, цитохимическое, иммунофенотипическое, цитогенетическое) исследование бластных клеток.
Цитогенетическое изучение выявило некоторые аномалии количества хромосом или их структуры в 90% случаев острого лимфобластного лейкоза. Гипердиплоидность (47 или более хромосом) выявлена у 1/3 больных. Обычно существуют хромосомные транслокации, вовлекающие протоонкогены; большинство из них демонстрируют транслокацию c-myc протоонкогена Хр8 в регион гена Хр14, кодирующий иммуноглобулины (Ig). - t (8; 14).
В случаях В-ОЛЛ встречаются t (2; 8) или t (8; 22). Это специфические транслокации, т.к. локус с c-myc дерегулируется промотором Ig. При пре-В-клеточном варианте острого лимфобластного лейкоза специфическая транслокация t (1; 19) наблюдается в 25% случаев. В результате гибридный ген Хр19 кодирует белок, который нарушает регуляцию транскрипции.
Описаны транслокации при Т-клеточном остром лимфобластном лейкозе, вовлекающие Хр11 и локус Т-клеточного рецептора (TCR) на Хр14 и Хр7, а также делеции Хр6, Хр9, Хр12, так что в этих случаях может происходить потеря опухолевых генов-супрессоров. Филадельфийская хромосома (Ph), t (9; 22) выявляется в 20% ОЛЛ у взрослых и менее чем в 5% у детей. Патологический белок, кодируемый гибридным геном при этой транслокации, при ОЛЛ отличается от белка, продуцируемого химерным геном BCR/ABL при хроническом миелолейкозе.
Лимфобласты при ОЛЛ всегда весьма гетерогенны.
Выделяют три подтипа острого лимфобластного лейкоза:
L1 - лимфобласты малой величины, имеют круглое или овальное ядро с тонко организованным хроматином, единичными нуклеолами и ободком голубой цитоплазмы.
L2 - лимфобласты средней и большой величины, имеют ядро неправильной формы с рыхлым хроматином, одну или более нуклеол и ободок умеренно серо-голубой цитоплазмы.
L3 - лимфобласты крупных размеров, с круглым или овальным ядром, содержащим глыбчатый хроматин, 2-5 нуклеол и ободок темно-голубой и частично вакуолизированной цитоплазмы.
С учетом иммунологических маркеров можно выделить четыре подтипа ОЛЛ, три из них В-клеточного и один - Т-клеточного происхождения.
Имеются два подтипа, состоящие из предшественников В-клеток:
- пре-пре-В клеточный ОЛЛ (морфологически - L1 вариант),
- пре-В клеточный ОЛЛ (морфологически - L2 вариант).
Третий подтип В-клеточного ОЛЛ состоит из более зрелых В-клеток (L3 вариант).
Около 20% случаев ОЛЛ происходят из предшественников Т-клеток и относятся морфологически к L2 варианту. Демонстрируется реаранжировка генов в- или гамма-цепей рецептора Т-клеток (TCR).
ОЛЛ из нуль-клеток - это малая пропорция случаев ОЛЛ, отличающаяся по маркерам от В- или Т-линии лимфоцитов и поэтому называемая нуль-ОЛЛ или неклассифицируемым ОЛЛ.
При цитохимическом исследовании характерно наличие терминальной динуклеотидил-трансферазы (Тс1Т) в 95% случаев пре-пре-В-, пре-В клеточного ОЛЛ и Т-клеточном ОЛЛ.
Активность фермента снижена в зрелых В-лимфоцитах и при L3 варианте ОЛЛ. При ОНЛЛ активность фермента выявляется только в 5-10% случаев.
Таблица 2. Характеристика бластов при ОЛЛ (М.А. Френкель, 2001)

Реакции на МПО, специфические и неспецифические эстеразы, ШИК-реакция - отрицательные. Реакция на окрашивание масляным красным - положительная.
Читайте также: