
Моносомия 17 хромосомы лейкоз
Кариотип клеток костного мозга у больных с миелодиспластическими синдромами (МДС) интенсивно изучается на протяжении последних 10—15 лет. Аномальные клоны выявлены до лечения у 30—50 % пациентов, в некоторых сообщениях приведены более высокие показатели — 60—75 %.
Обнаружение клонов клеток с аномальным кариотипом при миелодиспластическом синдроме (МДС) имеет важное теоретическое и клиническое значение, поскольку свидетельствует о принадлежности этой группы заболеваний к новообразованиям.
Цитогенетические изменения весьма разнообразны, спектр их близок к спектру хромосомных аномалий, наблюдаемых при остром нелимфобластном лейкозе, особенно вторичных.
Наиболее характерны моносомии 5 и 7, а также делеции длинного плеча этих хромосом, появление дополнительной хромосомы 8 и делеции длинного плеча хромосомы 20.
Установлено, что частота обнаружения клонов анеуплоидных клеток нарастает по мере прогрессирования болезни: на относительно ранних этапах она составляет 20—30 %, при появлении начальных признаков трансформации в острый лейкоз — до 40—60 %, при трансформации в острый миелобластный лейкоз — 80—90 %.
Транслокации, специфичные для первичных острых нелимфобластных лейкозах, при миелодисплазиях наблюдаются редко. Есть сообщения о повторяющихся транслокациях t(3;3)(q21;q26), t(8;21)(q22;q22) и t(3;21)(q26;q22). Примеры перестроек длинного плеча хромосомы 3 показаны на рисунке.
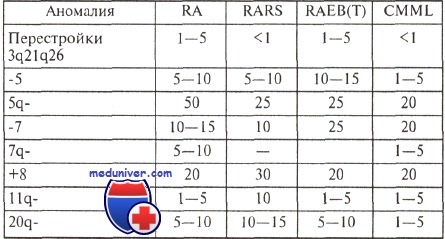
Основные хромосомные аномалии, характерные для миелодисплазий:
-7 или 7q-
-5 или 5q-
t(1;7)(q10;p10)
del(12)(р12-р13)
t(2;ll)(p13;q23)
del(13)(обязательно с включением 3q14)
t(6;9)(p23;q34)
del(20)(q11ql3)
+8
t(1;3)(p36;q21)
Перечисленные хромосомные аномалии наблюдаются при различных формах миелодисплазий, но частота их несколько различается.
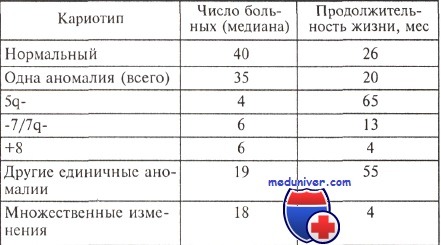
Опыт большинства исследователей свидетельствует о том, что существует корреляция между особенностями кариотипа и продолжительностью жизни больных с миелодиспластическим синдромом (МДС). Относительно благоприятным считается прогноз, если выявлены клоны клеток с единственной перестройкой 5q- или 20q; в то же время при любом варианте миелодиспластического синдрома обнаружение клона с множественными хромосомными аномалиями является крайне неблагоприятным.
Остановимся подробнее на отдельных нарушениях кариотипа, характерных для миелодиспластического синдрома (МДС).
Синдром 5q — рефрактерная сидеробластная анемия у пожилых больных, преимущественно женщин. В новой классификации ВОЗ этот синдром выделен как самостоятельный вариант миелодиспластического синдрома (МДС). Характерна макроцитарная анемия, резистентная к лечению, в костном мозге — признаки миелодисплазии клеток красного ряда и мегакариоцитов. Число тромбоцитов нормально или повышено, в костном мозге наблюдается гиперплазия гиполобулярных микромегакариоцитов. Клиническое течение сравнительно медленное. Трансформации в острый лейкоз наблюдаются приблизительно в 10 % случаев. Синдром впервые описан van den Berghe и соавт. в 1974—1985 гг..
Делеции длинного плеча хромосомы 5 наблюдаются и при других гематологических заболеваниях.
Предполагают, что делетирующийся участок содержит один или более генов-супрессоров. В этом направлении ведутся интенсивные исследования. До настоящего времени не подтверждена важная роль в патогенезе рефрактерной анемии ни одного из изучавшихся кандидатов на роль гена- супрессора.
Синдром моносомии хромосомы 7 встречается преимущественно у мальчиков до 4 лет. Характерна спленомегалия, часто наблюдаются лейкоцитоз с моноцитозом, тромбоцитопения, анемия. Прогноз плохой.
Как отмечалось, утрата одной из хромосом 7-й пары (моносомия 7) наблюдается при самых различных гемобластозах, включая острый нелимфобластный лейкоз, при этом она обычно ассоциирована с неблагоприятным прогнозом.
Делеции короткого плеча хромосомы 17 (17р-) обычно входят в число сложных изменений кариотипа. Как правило, 17р- сочетается с двумя или более хромосомными аномалиями и имеет неблагопрятное прогностическое значение.
В 75 % случаев в присутствии маркера 17р- наблюдается своеобразный дисгранулоцитопоэз в виде псевдопельгеровских гиподольчатых ядер и вакуолизации цитоплазмы. Этот маркер обнаружен не только при миелодисплазиях, но и при самых разнообразных злокачественных новообразованиях, включая солидные опухоли, его присутствие — плохой прогностический признак.
В 1997 г. опубликованы материалы международного совещания, посвященного диагностике и прогнозированию миелодиспластического синдрома. На основании ретроспективной оценки длительности заболевания до перехода в острый лейкоз и общей продолжительности жизни больных результат цитогенетического анализа был расценен как важнейший прогностический признак. В группу с благоприятным прогнозом отнесены случаи с единичными хромосомными аномалия ми: -Y, 5q- и 20q-. Неблагоприятное течение наблюдали при множественных (сложных) нарушениях (три или более перестройки кариотипа) и изменениях хромосомы 7 (делеции длинного плеча, моносомии).
Важное диагностическое значение может иметь метод FISH в тех случаях, когда стандартное цитогенетическое исследование неинформативно или обнаруживаются только единичные клетки с нарушением кариотипа, которое по формальным критериям нельзя считать клоном. Для диагностики наиболее характерных хромосомных нарушений при миелодиспластическом синдроме разрабатывается панель FISH-зондов.
Попытки выделить цитогенетические особенности каждой из клинико-морфологических субъединиц, входящих в общую гетерогенную группу миелодиспластических синдромов, не увенчались успехом. В то же время ХММЛ, рассматриваемый как миелопролиферативное заболевание с морфологическими признаками миелодисплазии, нередко ассоциируется со специфической хромосомной аномалией t(5;12)(q33;p13), однако в большинстве случаев ХММЛ эта хромосомная аномалия не выявляется.
Медицинский эксперт статьи

Исторически диагностика острого миелобластного лейкоза основана на цитоморфологии. Заболевание представляет собой морфологически гетерогенную группу.
В настоящее время общепризнана классификация по критериям FAB (French-American-British Cooperative Group). Основа этой классификации - соответствие морфологического субстрата лейкоза определённому ряду и уровню дифференцировки нормальных гемопоэтических клеток.
FAB-классификация острого миелобластного лейкоза
ОМЛ с минимальной дифференцировкой
Отсутствие созревания, активность миелопероксидазы менее 3%, есть иммунологические маркёры миелоидной дифференцировки
ОМЛ без созревания
Количество бластов превышает или равно 90% неэритроидных клеток, активность миелопероксидазы менее 3%
ОМЛ с созреванием
Более 10% миелоидных клеток имеют признаки созревания до промиелоцитов, количество моноцитов менее 20%
Острый промиелоцитарный лейкоз
Доминирующие клетки - промиелоциты с выраженной атипией
Острый промиелоцитарный лейкоз
Доминирующие клетки - промиелоциты с микрогрзкуляцией и резко положительной реакцией на миелопероксидазу
Острый миеломоноцитарный лейкоз
Количество миеломоноцитарных властных клеток с моноцитарным компонентом более 20% и менее 80%
Острый миеломоноцитарный лейкоз
Вариант М, с атипичными эозинофилами (>5%)
Острый монобластный лейкоз
Количество монобластов в костном мозге >80%
Острый монобластный лейкоз
Количество монобластов и моноцитов в костном мозге г80%
Острый эритроидный лейкоз
Доля эритробластов среди нуклеаров в костном мозге £50%, доля бластов среди неэритроидных клеток - более 30%
Острый мегакариоцитарный лейкоз
Морфологические черты мегакариобластов, CD4V, CD6V
Морфологические и иммунологические признаки
Морфологическая находка, высокоспецифичная для острого миелобластного лейкоза, - так называемые палочки Ауэра. Если реакция на миелопероксидазу отрицательна, что характерно для варианта М0, и обнаруживают палочки Ауэра, необходимо выставить диагноз острого лейкоза варианта М1. При вариантах М1 и М2 с t(8;21) часто наблюдают длинные нежные нитеподобные палочки Ауэра; при варианте М3 в цитоплазме можно увидеть пучки этих палочек.
Иммунологические признаки миелоидной дифференцировки включают нелинейные маркёры гемопоэтических предшественников CD34 и HLA-DR, панмиелоидные маркёры CD13, CD33 и CD65; маркёры, ассоциированные с моноцитами и гранулоцитами CD14 и CD15; линейные мегакариоцитарные маркёры CD41 и CD61; внутриклеточную миелопероксидазу.
Значение проточной цитофлюорометрии в диагностике острого миелобластного лейкоза существенно в случаях, когда необходима верификация вариантов М0 и М1, а также в диагностике бифенотипического лейкоза. Кроме того, метод позволяет разграничить варианты М0 и М1, а также варианты с гранулоцитарной дифференцировкой - М2 и М3.
Для определения стратегии лечения важно выделение так называемого острого бифенотипического лейкоза (biphenotypic acute leukemia, BAL). Критерии диагностики бифенотипического лейкоза основаны на оценке соотношения специфических лимфоидных и миелоидных маркёров, экспрессируемых властными клетками.
Значение современных лабораторных исследований в диагностике острого миелобластного лейкоза многократно возросло в течение двух последних десятилетий. Наибольшую важность приобрели цитогенетические характеристики, именно они признаны решающими прогностическими факторами. До начала 1990-х годов исследования проводили на клеточном уровне: оценивали структуру и число хромосом, наличие хромосомных аберраций в опухолевых клетках. Позднее исследования были дополнены молекулярно-биологическими методами, объектом изучения при этом стали химерные гены, появившиеся в результате аберраций хромосом, и белки - продукты их экспрессии. Цитогенетические изменения в лейкемических клетках обнаруживают у 55-78% взрослых пациентов и у 77-85% детей. Ниже приведено описание наиболее частых и значимых в клиническом отношении хромосомных аберраций при остром миелобластном лейкозе и их прогностическое значение.
Транслокация, ассоциированная с острым промиелоцитарным лейкозом, - t(15;17)(q22;ql2) с образованием химерного гена PML-RARa. Частота обнаружения этой аномалии составляет 6-12% всех случаев острого миелобластного лейкоза у детей, при варианте М3 она равна 100%. Транскрипт PML-RARa - маркёр лейкемии, то есть у пациентов, достигших ремиссии, его не обнаруживают, а повторное его выявление во время морфологической ремиссии - предвестник клинического рецидива острого промиелоцитарного лейкоза.
Инверсия хромосомы 16 - inv(16)(pl3;q22) - и её вариант t(16;16) характерны для миеломоноцитарного лейкоза с эозинофилией М4Е0, хотя наблюдаются и при других вариантах острого миелобластного лейкоза.
Реаранжировка 1 Iq23/MLL. Регион 23 длинного плеча одиннадцатой хромосомы достаточно часто становится участком структурных реаранжировок у детей с острым лейкозом - как лимфобластным, так и миелобластным. При первичном остром миелобластном лейкозе аномалию llq23 обнаруживают у 6-8% больных. при вторичном - у 85%, что связано с воздействием эпиподофиллотоксинов - ингибиторов топоизомеразы.
Инверсия inv(3)(q21q26)/t(3;3)(q21;q26) описана при всех вариантах острого миелобластного лейкоза, за исключением M3/M3v и М4Е0. Несмотря на отсутствие связи между определённым FAB-вариантом и инверсией хромосомы 3, у большинства больных в костном мозге обнаруживают общие морфологические признаки: увеличение числа мегакариоцитов и многочисленные микромегакариоциты.
Транслокация t(6;9)(p23;q34) описана более чем у 50 пациентов с острым миелобластным лейкозом. В большинстве случаев это единственная хромосомная аномалия. Несколько чаще t(6;9) выявляют у пациентов с М2 и М4 вариантами, хотя встречается она при всех формах острого миелобластного лейкоза.
Транслокация t(8;16)(pll;pl3) описана у 30 больных острым миелолейкозом, преимущественно при вариантах М4 и М5. Чаще аномалию обнаруживают у юных пациентов, в том числе у детей до года.
Моносомия (-5) и деления del(5)(q-). Потеря участка длинного плеча или всей пятой хромосомы не связана с каким-либо определённым вариантом острого миелобластного лейкоза. Часто это дополнительная аномалия при сложных аберрациях.
Моносомия (-7) и деления del(7)(q-). Моносомия в седьмой паре хромосом - вторая по частоте, после трисомии (+8), аберрация среди количественных транслокаций (то есть транслокаций, изменяющих число хромосом).
Трисомия (+8) - наиболее частая количественная аберрация, составляющая 5% всех цитогенетических изменений при остром миелобластном лейкозе.
Делеция del(9)(q-). Потеря длинного плеча девятой хромосомы часто сопровождает благоприятные аберрации t(S;21), реже inv(16) и t(15;17), не оказывая влияния на прогноз.
Трисомия (+11), подобно другим трисомиям. может быть солитарной аномалией, но чаще встречается совместно с другими численными или структурными хромосомными аберрациями.
Трисомия (+13) в 25% бывает солитарной аберрацией, чаще наблюдаемой у пациентов в возрасте 60 лет. Она ассоциирована с хорошим ответом на терапию, однако рецидивы отмечаются часто и общая выживаемость невысока.
Трисомия (+21). Данную аномалию обнаруживают у 5% больных острым мислобластным лейкозом, менее чем в 1% случаев она является солитарной. Связи с каким-либо вариантом FAB не обнаружено.
Помимо перечисленных выше, существуют описанные у очень небольшого числа пациентов транслокации, роль которых в развитии заболевания и прогностическое значение не ясны. Это количественные аберрации четвёртой, девятой и двадцать второй пар хромосом, а также структурные транслокации t(l;3) (p36;q21). t(l;22)(pl3;ql3), t(3;21)(q26;q22), t(7;ll)(pl5;pl5). t(ll;17)(q23;q25) и t(16;21)(pll;q22).

[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20]
Полный текст:
- Аннотация
- Об авторах
- Список литературы
- Cited By
1. Nybakken G E, Bagg A. The genetic basis and expanding role of molecular analysis in the diagnosis, prognosis, and therapeutic design for myelodysplastic syndromes. J Mol Diagn. 2014 Mar;16(2):145-58
2. Arber D, Orazi A, Hassrjian R. The 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20).
4. Greenberg P, Cox C, LeBeau MM, Fenaux P et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997 Mar 15;89(6):2079-88.
5. Greenberg P L, Tuechler H, Schanz J Et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-65. doi: 10.1182/blood-2012-03-420489.
6. Zemanova Z, Michalova K, Buryova H Et al. Involvement of deleted chromosome 5 in complex chromosomal aberrations in newly diagnosed myelodysplastic syndromes (MDS) is correlated with extremely adverse prognosis. Leuk Res. 2014 May;38(5):537-44
7. Wang S A, Abruzzo L V, Hasserjian R P et al. Myelodysplastic syndromes with deletions of chromosome 11q lack cryptic MLL rearrangement and exhibit characteristic clinicopathologic features. Leukemia Research 35 (2011) 351-357.
8. Ferrara F, Schiffer C A. Acute myeloid leukemia in adults. Lancet. 2013 Feb 9;381(9865):484-95.
9. Breems D A, Van Putten W L et al. Monosomal Karyotype in Acute Myeloid Leukemia: A Better Indicator of Poor Prognosis Than a Complex Karyotype. J Clin Oncol. 2008 Oct 10;26(29):4791-7. doi: 10.1200/JCO.2008.16.0259.
10. Renneville A, Roumier C, Biggio V Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008 May;22(5):915-31.
11. Liehr T. Fluorescence In Situ Hybridization (FISH) -Application Guide. Springer-Verlag Berlin Heidelberg. 2009, p I (16).
12. Pinheiro R F, Chauffaille M L. Comparison of I-FISH and G-banding for the detection of chromosomal abnormalities during the evolution of myelodysplastic syndrome. Braz J Med Biol Res. 2009; 42(11):1110-2.
13. Malcovati L, Hellstrоm-Lindberg E, Bowen D et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943-64.
14. Speicher M R, Gwyn Ballard S, Ward D C. Karyotyping human chromosomes by combinatorial multi- fluor FISH. Nat Genet. 1996 Apr;12(4):368-75.
15. Schrоck E, du Manoir S, Veldman T et al. Multicolor spectral karyotyping of human chromosomes. Science. 1996; 273(5274):494-7.
16. Shaffer L G, McGowan-Jordan J, Schmid M. ISCN: An International System for Human Cytogenetic Nomenclature, S. Karger, Basel, Switzerland, 2013.
17. Fenaux P. Myelodysplastic syndromes: from pathogenesis and prognosis to treatment. Semin Hematol. 2004 (2 Suppl 4):6-12.
18. Navada S, Chatalbash A, Silverman L. Clinical significance of cytogenetic manifestations in myelodysplastic syndromes. LabMedicine, 2013; 44: 103-107.
19. Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol. 2008; 87(7): 515-26. doi: 10.1007/s00277-008-0483-y.
20. List A, Kurtin S, Roe D J Et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005; 352(6):549-57.
21. Fenaux P, Mufti G J, Hellstrom-Lindberg E et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomized, open-label, phase III study. Lancet Oncol. 2009;10(3):223-32.
22. Volkert S, Kohlmann A, Schnittger S et al. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer. 2014 May;53(5):402-10.
23. Tanke H J, Wiegant J, van Gijlswijk RP et al. New strategy for multi-color fluorescence in situ hybridization: COBRA: combined binary ratio labelling. Eur J Hum Genet. 1999; 7(1):2-11.
24. Babicka L, Ransdorfova S, Brezinova J et al. Analysis of complex chromosomal rearrangements in adult patients with MDS and AML by multicolor FISH. Leukemia Research; 31(2007): 39-47.
25. Mrоzek K. Cytogenetic, molecular genetics, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol. 2008 Aug;35(4):365-77.
26. Andersen M, Christiansen D et al. Duplication or amplification of chromosome band 11q23, including the unrearranged MLL gene, is a recurrent abnormality in therapy-related MDS and AML, and is closely related to mutation of the TP53 gene and to previous therapy with alkylating agents. Genes Chromosomes Cancer. 2001; 31(1):33-41.
27. Sarova I, Berezinova J et al. Characterization of Chromosome 11 Breakpoints and the Areas of Deletion and Amplification in Patients with Newly Diagnosed Acute Myeloid Leukemia. Genes Chromosomes Cancer. 2013; 52:619-635
28. Tang G, DiNardo CD, Zhang L. et al. MLL gene amplification in acute myeloid leukemia and myelodysplastic syndromes is associated with characteristic clinicopathological findings and TP53 gene mutation. Hum Pathol 46(1):65-73, 1/2015. e-Pub 10/2014. PMID: 25387813.

Контент доступен под лицензией Creative Commons Attribution 4.0 License.

Для того чтобы повысить вероятность эффективного лечения опухоли, необходимо выявить ее как можно раньше. При выявлении опухоли определяют стадию ее развития. Процесс определения распространенности процесса называют стадированием. Как правило, здесь основную роль играют размеры опухоли и ее распространенность по организму.
Что касается лейкозов, то процесс их стадирования не похож на процесс стадирования других видов рака. Особенность лейкоза – в поражении костного мозга и периферической крови. В результате этого лейкозные клетки проникают в печень, селезенку, лимфатические узлы, яички, яичники или ЦНС.
Большое количество проникнутых в ЦНС клеток можно увидеть при помощи исследования спинномозговой жидкости под микроскопом. Для этого делают спинномозговую (люмбальную) пункцию в качестве составной части первичного обследования.
Когда лейкозные клетки были обнаружены, нужно начать их интенсивное лечение.
Больных делят на пациентов с хорошим и плохим ответом на лечение. Различия, которые между ними существуют, называют прогностическими факторами. Они играют весомую роль в определении прогноза заболевания и интенсивности терапии.
Прогностические факторы являются более важными и значительными для больных ОЛЛ, чем ОМЛ.
Детей с диагнозом ОЛЛ подразделяют на несколько категорий: низкого риска, стандартного риска, высокого риска и очень высокого риска. Степень интенсивности терапии зависит от категории. Чем она выше, тем высшую терапию нужно использовать.
Как правило, дети с низким риском могут рассчитывать на положительные результаты лечения больше, чем больные с очень высоким риском.
Одним из важных прогностических факторов является возраст больного и исходное количество лейкоцитов. При этом не стоит забывать того, что даже при наличии одного или более неблагоприятных прогностических факторов, положительные результаты возможны.
Если на момент диагностики возраст ребенка составляет от одного года до девяти лет, прогноз заболевания будет лучшим. У больных в возрасте до года и от десяти лет, прогноз относят к категории высокого риска.
Больные ОЛЛ, у которых содержание лейкоцитов находиться на очень высоком уровне (более 50000 клеток в куб.мм.) входят в категорию высокого риска, их нужно лечить с помощью применения более интенсивной терапии.
Возможность получения положительных результатов у девочек, больных ОЛЛ, несколько выше, чем у мальчиков.
Выздоравливание афроамериканских и латиноамериканских детей, страдающих ОЛЛ, происходит реже в сравнении с другими расами.
Дети, которым был поставлен пре-В или ранний пре-В-клеточный ОЛЛ прогноз, выздоравливают лучше, чем дети с Т-клеточным и зрелым В-клеточным лейкозом (типа Беркитта).
Вероятность получения положительных результатов увеличивается у пациентов, с повышенным содержанием хромосом в лейкозных клетках. Особенно при наличии дополнительной хромосомы 4 или 10. Дети, у которых количество хромосом меньше нормы, имеют более низкую вероятность выздоровления.
Дети с транслокацией между хромосомами 12 и 21 дают более позитивные прогнозы, чем те, у кого транслокация между хромосомами составляет 9 и 22, 1 и 19 или 4 и 11.
Больные, которые находятся в периоде ремиссии (отсутствие признаков заболевания) на протяжении 7-14 дней химиотерапии, дают более позитивные прогнозы по сравнению с остальными детьми с ОЛЛ, которые нуждаются в более интенсивном лечении.
У детей, больных ОМЛ, прогностические факторы не являются такими важными, как у детей больных ОЛЛ.
У детей с диагнозом ОМЛ и количеством лейкоцитов менее 100000 в куб.мм. на момент диагностики, положительные результаты более вероятны, чем у больных с более высоким количеством лейкоцитов.
Миелодиспластический синдром или вторичный ОМЛ. Больные, перенесшие миелодиспластический синдром или ОМЛ, возникший в результате лечения другой опухоли, дают менее благоприятные прогнозы.
У больных ОМЛ, с дефектом хромосом под названием моносомия 7, прогноз заболевания негативный. Дети, у которых транслокации между хромосомами составляют 15 и 17, 8 и 21 или у которых инверсия хромосомы находиться на отметке 16, имеют больше шансов на выздоровление.
От морфологии (вида лейкозной клетки под микроскопом) зависит степень выживаемости больных. Внутри некоторых клеток у больных ОМЛ находятся специальные гранулы (тельца Ауэра). Самое большое их количество наблюдается при м2 и м3 типах ОМЛ. Все это дает благоприятные прогнозы.
Дети, больные синдромом Дауна имеют большую вероятность на излечение от ОМЛ, чем дети, не имеющие этого генетического заболевания.
Больные ОМЛ, не отреагировавшие на терапию (достижение ремиссии после одного цикла химиотерапии), имеют большую вероятность излечения в сравнению с теми, кто нуждался в более длительной терапии или теми, у кого вообще не наблюдалась реакция на лечение.
При лечении острого лимфобластного лейкоза выделяют три фазы: индукцию, консолидацию или интенсификацию, а также поддерживающую фазу.
Индукция. Цель индукции – достичь ремиссии. Это значит, что должны исчезнуть лейкозные клетки из периферической крови и костного мозга и восстановиться нормальные показатели кроветворения.
Через месяц лечения у 95% детей достигается полная ремиссия. При этом лечение должно быть весьма интенсивным, его также могут сопровождать инфекционные осложнения. Стоит отметить, что смертность больных от таких осложнений составляет около 3%.
Во время диагностики ОЛЛ в организме больного насчитывают более 100 миллиардов опухолевых клеток. Чтобы достичь ремиссии, нужно уничтожить 99,9% лейкозных клеток. Однако в организме ребенка все еще остается более 100 млнлейкозных клеток, которые также необходимо истребить.
В связи с этим, больной в течение 4-6 месяцев проходит фазу консолидации, а после этого – поддерживающую терапию в течение двух лет.
Больных со стандартным риском на протяжении месяца лечат при помощи химиотерапии с использованием трех препаратов (аспарагиназа, винкристина и преднизолона или дексаметазона).
Более того, в профилактических целях, в спинномозговой канал вводят метотрексат. Детям с высоким риском кметотрексату добавляют цитарабин и гидрокортизон. Эти же больные могут подвергаться облучению головного и, возможно, спинного мозга.
Консолидация. Длительность этой, более интенсивной степени - 4-8 месяцев. Дети со стандартным риском употребляют метотрексат и 6-меркаптопурин или 6-тиогуанин. К этим препаратам также могут добавить винкристин и преднизолон.
Больные с высоким риском дополнительно употребляют аспарагиназ, доксорубицин, циклофосфамид и цитарабин.
Поддерживающее лечение назначают после того, как завершиться фаза индукции и консолидации. Как правило, больным внутрь вводят метотрексат и 6-меркаптопурин, а также преднизолон или дексаметазон. Больным с высоким риском назначают более интенсивное лечение.
Общая длительность всего лечения составляет несколько лет.
На тактику лечения влияет срок наступления рецидива заболевания. Чем раньше он наступил, тем интенсивнее должна быть терапия.
Больные с ОЛЛ низкого риска имеют вероятность излечения 85-95%, стандартного риска - 65-85% и высокого риска - 60-65%.
Моносомия относится к генетическим аномалиям, для которых характерно изменение кариотипа. В норме у человека определяется 23 хромосомы, каждая из которых имеет гомологичную пару. Если одна из них лишается своей пары, то развивается моносомия. Заболевание протекает тяжело, часто приводит к внутриутробной гибели плода. В других случаях ребенок рождается живым, но при этом наследует тяжелые врожденные изменения, которые объединяют в синдромы, самыми распространенными из которых являются синдром Шерешевского-Тернера и кошачьего крика.
Синдром Шерешевского-Тернера
Является следствием моносомии по хромосоме Х. Больные дети часто рождаются недоношенными или имеют сниженную массу тела. Одним из классических признаков синдрома Шерешевского-Тернера, который можно заметить сразу после рождения, является выраженная кожная складка на шее. Среди других клинических проявлений отмечаются:
- пороки сердца;
- отечность верхних и нижних конечностей;
- нарушение циркуляции лимфы;
- задержка речевого и физического развития.
По мере взросления ребенка проявляются характерные черты строения тела. Рост обычно не превышает 150 см, крыловидные складки на шее сохраняются, ушные раковины могут деформироваться, верхняя челюсть недоразвита, грудная клетка широкая. Моносомия по хромосоме Х влияет на развитие органов половой системы. У женщин отмечается отсутствие фолликулов в яичниках, нарушение менструального цикла, недоразвитие молочных желез. У мужчин снижается уровень тестостерона, может отсутствовать одно или оба яичка либо отмечаться их недоразвитие.
Прогноз при синдроме Шерешевского-Тернера относительно благоприятный. При отсутствии тяжелых пороков развития и регулярном наблюдении у специалиста продолжительность жизни не сокращается.
Синдром кошачьего крика
Является примером частичной моносомии. В данном случае теряется не вся хромосома из одной пары, а только определенный участок — короткое плечо 5-й хромосомы. Заболевание получило свое название из-за специфического плача, который издают новорожденные дети. Он обусловлен недоразвитием гортани и ее хрящевых компонентов. Кроме того, выявляются и другие симптомы данного вида моносомии:
- отклонение в умственном и физическом развитии;
- изменение формы головы и черт лица (недоразвитая нижняя челюсть, специфический внешний вид глаз и ушных раковин);
- недостаток массы тела;
- врожденные пороки развития (микроцефалия, пороки сердца, мышц, внутренних органов).
Как и предыдущий вид моносомии, синдром кошачьего крика характеризуется благоприятным течением при условии, что отсутствуют тяжелые пороки, которые могут стать причиной летального исхода.
Причины моносомии
Моносомия может возникать на различных стадиях клеточного деления. Например, при синдроме Шерешевского-Тернера нарушается процесс расхождения Х-хромосом. В результате в одну яйцеклетку женщины попадает две Х-хромосомы, а во вторую ни одной. Во время процесса оплодотворения зигота получает набор Х0 и Y0,- вместо нормального ХХ или XY.
Причины появления моносомии не связаны с наследственными факторами. Нарушения возникают при воздействии неблагоприятных факторов. Оказывать влияние на половые клетки могут вредные привычки, радиация, некоторые лекарственные препараты, химические вещества, неблагоприятная экологическая обстановка, вредные условия труда и т. д.
Диагностика моносомии
Выявить заболевание можно еще на этапе внутриутробного развития. Для этого всем беременным женщинам проводится скрининговое УЗИ. Если специалист выявляет нарушения развития плода, то дополнительно назначается биопсия хориона, которая позволяет получить образец ткани и определить кариотип. Таким способом можно выявить не только моносомии, но и другие генетические нарушения.
Читайте также: