
Миелопролиферативное заболевание с гиперэозинофилией
Гиперэозинофильный синдром (ГЭС) — это гетерогенная группа заболеваний, для которых характерны повышенный уровень эозинофилов в крови и повреждение внутренних органов. Первое упоминание о злокачественной эозинофилии принадлежит Stillman, который в 1912 г. описал группу больных с выраженной эозинофилией в крови и тяжелым течением заболевания.
Выделяют три основные группы заболеваний, протекающих с эозинофилией:
1) реактивные (неклональные) эозинофилии;
2) клональные заболевания кроветворной системы;
3) идиопатический гиперэозинофильный синдром (диагноз ставят после исключения первых двух заболеваний).
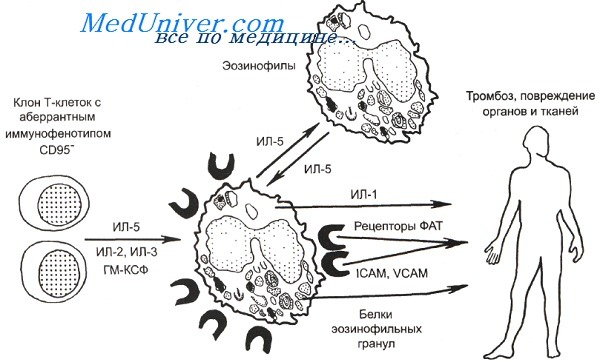
Патогенез лимфопролиферативного варианта гиперэозинофильного синдрома.
ФАТ — фактор активации тромбоцитов; IСАМ — молекулы межклеточной адгезии; VCAM — молекулы сосудистой адгезии.
Эозинофилии 1-й группы неклонального генеза (симптоматические, реактивные) наблюдаются при следующих заболеваниях:
• гельминтозах;
• инфекциях: бактериальных, грибковых, вирусных;
• аллергических заболеваниях (астма, ринит, атопический дерматит, крапивница, экзема, пищевая и лекарственная аллергия);
• заболеваниях дыхательных путей (синдром Лефлера, бронхоэктатическая болезнь, муковисцидоз);
• заболеваниях соединительной ткани (ревматоидный артрит, гранулематоз Вегенера, узелковый полиартериит и др.);
• злокачественных заболеваниях (болезнь Ходжкина, опухоли ЖКТ, рак молочной железы, опухоли почек, опухоли легких и др.).
При аллергических состояниях и инфекциях активируются Th2-клетки, продуцирующие ИЛ-5. Кроме того, активированные эозинофилы способны к аутокринной стимуляции путем секреции ИЛ-1, ИЛ-5, TGFa и PAF.
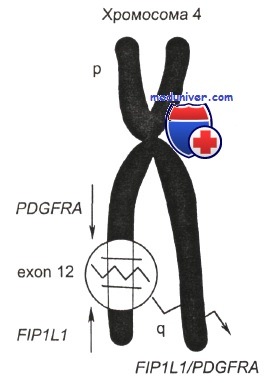
Хромосомная поломка при миелопролиферативном варианте гиперэозинофильного синдрома
Вторую большую группу эозинофилии представляют эозинофилии, сопровождающие клональные заболевания кроветворной ткани. При гемобластозах эозинофилы могут быть частью злокачественного клона: например, эозинофилы при Ph-позитивном хроническом миелолейкозе. В других случаях лейкозные клетки могут продуцировать цитокины, стимулирующие пролиферацию нормальных эозинофилов. Dombret назвал такие эозинофилии параклональными. В современной классификации опухолей гемопоэтической и лимфоидной тканей ВОЗ (2001) выделены хронический эозинофильный лейкоз (ХЭЛ)/гиперэозинофильный синдром (ГЭС), относящийся к группе миелопролиферативных заболеваний, и острый миеломонобластный лейкоз (ОММЛ) с увеличенным количеством эозинофилов костного мозга с inv(16)(p13;q22) или t(16;16)(p13;q22) (CBFb/MYH11).
Кроме этих двух заболеваний, эозинофилии могут сопровождать еще ряд гемобластозов:
1) хронический миелолейкоз (ХМЛ);
2) атипичный ХМЛ;
3) миелодиспластические синдромы;
4) острый лимфобластный лейкоз;
5) Т-лимфобластную лимфому;
6) системные заболевания тучных клеток.
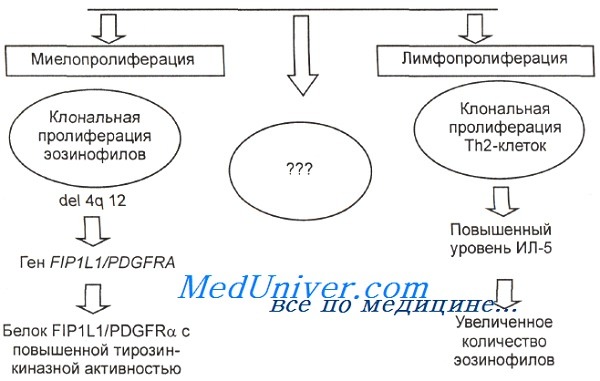
Этиология идиопатического гиперэозинофильного синдрома.
Тем не менее у многих больных даже после тщательного поиска не удается найти какой-либо известной причины эозинофилии. В этих случаях может быть диагностирован идиопатический ГЭС.
Этиология гиперэозинофильного синдрома до сих пор полностью не ясна. Предполагают, что гиперэозинофильный синдром является гетерогенной группой заболеваний, в основе которой лежит либо вторичная эозинофилия, вызванная гиперпродукцией эозинофилотропных цитокинов клональной популяцией лимфоцитов, либо это первичное миелопролиферативное заболевание. Признаки клональности удается обнаружить не всегда даже с помощью современных методов диагностики. Многие авторы высказывают предположение, что в основе гиперэозинофильного синдрома лежит увеличенная пролиферация Т-клеток, секретирующих ряд цитокинов. В 1993 г. Н. Suzushima и соавт. у пациента с типичной клинической картиной гиперэозинофильного синдрома выявили цитогенетические поломки в популяции клеток, имеющих фенотип CD3+, CD4-, CD8.
Позже Е. Cogan и соавт. сообщили о клональной пролиферации Т-хелперов типа 2, секретирующих ИЛ-5, у больного гиперэозинофильным синдромом. Далее появились работы, доказывающие, что у некоторых пациентов с необъяснимой эозинофилией удается обнаружить популяцию Т-клеток с аберрантным иммунофенотипом или имеющих явные цитогенетические поломки. В последующем у отдельных больных развивались лимфопролиферативные заболевания. Косвенным подтверждением роли Т-хелперов в развитии гиперэозинофильного синдрома может быть эффективное лечение циклоспорином А и хлордеоксиаденозином (2CDA), который, как известно, подавляет пролиферацию Т-хелперов. Возможно, у части больных с гиперэозинофильным синдромомна момент диагностики патологический Т-клеточный клон не удается выявить доступными методами.
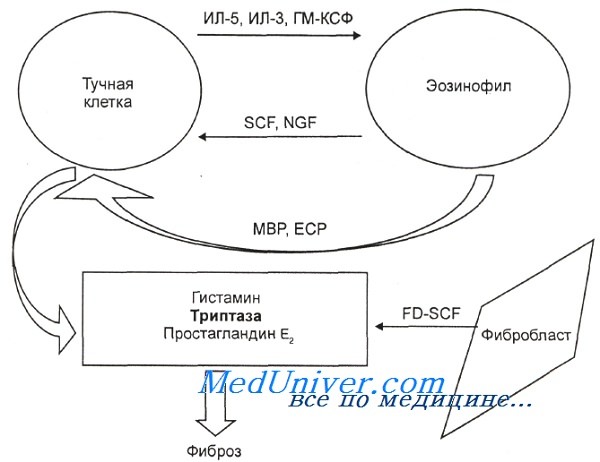
Схема развития фиброза при миелопролиферативном варианте гиперэозинофильного синдрома. SCF — фактор стволовых клеток; NGF — фактор роста нейронов; МВР — большой основной протеин, или БОП; ЕСР — эозинофильный катионный протеин, или ЭКП; FD-SCF — фибробластный фактор стволовых клеток.
В дальнейшем у этой категории больных по мере прогрессирования болезни и выявления признаков лимфопролиферации диагноз гиперэозинофильного синдрома может быть пересмотрен.
Открытие реаранжировки позволило не только получить новые сведения об этиологии гиперэозинофильного синдрома, который может теперь рассматриваться как миелопролиферативное заболевание по крайней мере у части больных, но и объясняет эффективность лечения больных гиперэозинофильным синдромом гливеком.
Независимо от этиологических факторов отсутствие адекватного лечения гиперэозинофильного синдрома (ГЭС) ведет к фатальному повреждению жизненно важных внутренних органов.

В норме в губчатом веществе костного мозга, находящегося в полостях крупных костей скелета, продуцируются незрелые стволовые клетки. Постепенно они созревают и преобразуются в полноценные форменные элементы:
- эритроциты, доставляющие кислород к органам и тканям,
- лейкоциты, защищающие организм от инфекционных агентов и прочих чужеродных веществ,
- тромбоциты, формирующие кровяной сгусток и останавливающие кровотечение.

созревание форменных элементов крови в норме
При наличии у человека миелопролиферативного заболевания в кровь поступают клетки, неспособные выполнять свои функции. Стволовые клетки при патологии часто преобразуются только в один тип форменных элементов. Патологический процесс отличается медленным прогрессированием.
Миелопролиферативное заболевание — это собирательное понятие, включающее группу гемобластозов, которые характеризуются аномальным разрастание костномозговых структур, отвечающих за образование кровяных телец. Выделяют несколько основных форм МПЗ, при которых поражаются разные клеточные элементы крови:
- Истинная полицитемия,
- Эссенциальная тромбоцитемия,
- Хронический миелолейкоз.
Причины

пример jak2 мутации при истинной полицитемии (избытке эритроцитов)
В основе МПЗ лежит приобретенная генная мутация, обусловленная влиянием негативных внешних или внутренних факторов. Мутация генов MPL и jak 2 приводит к повреждению ДНК одной гемопоэтической клетки, которая дает начало всем типам клеточных элементов. Аномально изменившаяся бластная форма приобретает отрицательные черты — перестает развиваться, не созревает полностью, не самоуничтожается, а непрерывно делится и порождает многочисленные клоны. Именно поэтому МПЗ называют клональным. Клоны также остаются на начальном уровне развития и имеют полностью недифференцированную структуру. Повреждаться может как один, так сразу несколько ростков кроветворения.
В результате в костном мозге увеличивается количество клеток-предшественниц эритроцитарного, тромбоцитарного и лейкоцитарного типов. По мере их накопления в кровяном русле ухудшается самочувствие больных. От того, какой росток переродился, зависит характер патологии, ее симптоматика и прогноз. Формы МПЗ отличаются медленным развитием. Если заболевание было выявлено на ранней стадии, у больного есть все шансы добиться стойкой ремиссии.
Причины, вызвавшие мутационные процессы, остаются до конца неизученными. Одни ученые относят к ним негативные факторы окружающей среды, другие — ошибки при делении клеток. МПЗ не является наследственным. Мутации генов могут возникать на протяжении всей жизни человека. Они называются приобретенными. Риск развития патологии увеличивается с возрастом. Лицам старше 50 лет необходимо внимательно относиться к здоровью и при появлении подозрительной симптоматики обращаться к гематологу. Вероятность развития недуга повышается под воздействием факторов риска — облучения и химикатов, оказывающих токсическое влияние на организм.
Классификация
Миелопролиферативные заболевания имеют код по МКБ 10 — D47.1. По типу течения их подразделяют на острые и хронические. В первую группу входят максимально агрессивные и быстро прогрессирующие недуги, поражающие в основном молодых людей. К группе хронических миелопролиферативных заболеваний относятся медленно развивающиеся патологии, имеющие относительно благоприятный прогноз и возникающие у пожилых лиц.
В зависимости от пораженного ростка кроветворения выделяют следующие формы процесса:
- Истинная полицитемия — гиперпродукция красных кровяных телец и сгущение крови. Эритроциты задерживаются в селезенке, развивается спленомегалия. У больных возникают признаки тромбогеморрагического синдрома, повышается риск инсультов и инфарктов. В целом данная форма отличается доброкачественным течением. По сравнению с другими видами МПЗ она характеризуется высокой выживаемостью.
- Эссенциальный тромбоцитоз — опасное для жизни состояние, при котором происходит усиленное образование тромбоцитарных клеток.
- Хронический миелолейкоз — злокачественное заболевание, характеризующееся преимущественным поражением гранулоцитарного ростка и появлением в крови недифференцированных лейкоцитов.
- Эозинофильная лейкемия — усиленное разрастание и повреждение эозинофилов, которые относятся к лейкоцитарным клеткам. При этом нарушаются их главные функции — борьба с инфекцией и иммунный ответ на потенциальные аллергены.
- Миелофиброз — образование в косном мозге патологически измененных клеток с заменой функциональной ткани соединительнотканными волокнами.
- Хронический нейтрофильный лейкоз — формирование незрелых нейтрофилов, которые перестают защищать организм от патогенов.

Классификация МПЗ имеет важное значение для диагностики онкологических заболеваний органов кроветворения. С ее помощью гематологи-онкологи могут легко определить тип сформировавшейся патологии и подобрать больному адекватную терапию, которая может спасти жизнь.
Видео: лекция по классификации и патогенезу ХМПЗ
Развитие и симптомы
Существует три пути распространения заболевания по организму:
- Лимфогенный — аномальные структуры проникают во внутренние органы по лимфатическим сосудам.
- Гематогенный — проникновение видоизмененных клеток в здоровые ткани по кровеносному руслу.
- Имплантационный — прорастание пораженных бластных форм в соседние органы и близлежащие ткани.
Гематогенное распространение злокачественных клеток считается самым опасным. Таким пациентам вместе с лечебными мероприятиями проводят динамическое наблюдение за функционированием внутренних органов. Данный тип патологии дает метастазы в самые отдаленные участки организма человека, что приводит к формированию вторичных онкологических очагов.
Клиническая картина МПЗ зависит от конкретной формы процесса, сопровождающегося разрастание кроветворных тканей костного мозга и чрезмерным поступлением в кровоток остановившихся в своем развитии атипичных кровяных телец. Каждый вид заболевания отличается характерной симптоматикой. Но существуют общие распространенные симптомы. Это признаки анемии или тромбоза:
- не проходящая слабость, быстрая утомляемость, упадок сил,
- отсутствие аппетита и похудание,
- шум в ушах и головокружение,
- помрачение сознания,
- дезориентация во времени и пространстве,
- гематомы на теле,
- частые кровотечения и кровоизлияния,
- отечность тканей и артралгия,
- абдоминальная боль,
- бледность кожи,
- гепатоспленомегалия,
- плетора (“полнокровие”),
- лихорадка.
Это общая симптоматика, возникающая при любой форме МПЗ. Существуют также специфические проявления, характерные для каждой из них.
-
Признаки, типичные для полицитемии: гепатомегалия и спленомегалия, гиперемия кожи, гипертензия, ночная потливость, головная боль, кожный зуд, диплопия, нечеткость зрения, онемение и жжение в ступнях, распирание и тяжесть в левом подреберье.


картина крови при тромбоцитемии

общая клиника лейкозов
Диагностика
Симптоматика МПЗ — основание для назначения пациенту диагностических процедур, позволяющих подтвердить или опровергнуть наличие процесса, а также выяснить, в какой именно форме протекает патология органов кроветворения.
Обследование начинают с опроса и сбора анамнеза. Врачи уточняют, какой образ жизни ведет больной, имеет ли пагубные пристрастия, какие заболевания перенес и чем лечился. Осмотр пациента – определение общего состояния и выявление признаков, которые обычно отсутствуют у здоровых людей.

Лабораторная диагностика МПЗ заключается в проведении целого ряда исследований и испытаний:
- Гемограмма — подсчет лейкоцитарной формулы, определение количества эритроцитов, тромбоцитов, уровня гемоглобина, гематокрит.
- Микроскопия мазка периферической крови — обнаружение бластных форм.
- БАК — определение функционального состояния печени и других внутренних органов.
- Миелограмма – результат микроскопии мазка пунктата костного мозга, отражающий качественный и количественный состав ядросодержащих клеток миелоидной ткани.

пункция КМ для миелограммы
Помимо лабораторной диагностики для постановки диагноза необходимы результаты инструментальных исследований. Больным проводят УЗИ брюшной полости для определения степени гепатоспленомегалии. В диагностически сложных случаях их направляют на томографическое исследование.
Лечение
Онкогематологи назначают лечение своим больным по результатам диагностических исследований. Существуют стандартные терапевтические методики, которые применяют при различных видах МПЗ. Если у пациента обнаружена начальная стадия процесса, когда еще отсутствуют клинические признаки, за ним устанавливают динамическое наблюдение. При появлении первых признаков патологии переходят непосредственно к лечению.

Каждому больному подбирается индивидуальная лечебная методика в соответствии с его состоянием и степенью выраженности имеющихся нарушений.

трасплантация КМ – наиболее радикальная, но и потенциально действенная методика при удачном исходе
После проведения полного лечебного курса наступает период реабилитации. Больной должен находится под постоянным наблюдением доктора и строго выполнять все его предписания, позволяющие организму быстрее восстановиться.
- правильное, сбалансированное питание с ограничением жирных, соленых, острых блюд и полным исключением алкоголя, курения;
- долгие пешие прогулки на свежем воздухе, желательно около водоема;
- исключение чрезмерного физического перенапряжения;
- соблюдение режима дня – полноценный сон, чередование труда и отдыха.
Миелопролиферативное заболевание — рецидивирующий процесс, способный обостриться в любое время. Именно поэтому всем пациентам необходимо регулярно посещать лечащего врача и проходить диагностические исследования с профилактической целью.
Прогноз МПЗ считается благоприятным только в случае успешной трансплантации костного мозга, которая разрешена не всем больным. Хронические формы переносятся легче острых. Продолжительность жизни пациентов в этом случае составляет 5–7 лет при условии получения комплексной терапии. Если у больных обнаружены метастазы, прогноз становится неутешительным — они погибают в течение 6 месяцев.
Видео: лекция об опыте лечения ХМПЗ
Миелопролиферативное заболевание это патология, характеризующаяся избыточной выработкой компонентов крови. У людей с такой болезнью увеличивается риск тромбозов. Чаще этому заболеванию подвержены мужчины старше 40 лет, реже женщины, а среди детей встречаются одиночные случаи.
- Виды и стадии патологии
- Симптоматика заболевания
- Диагностические мероприятия
- Методы лечения
Виды и стадии патологии
Существует несколько видов миелопролиферативных заболеваний:
- Эссенциальный тромбоцитоз характеризуется переизбытком тромбоцитов.
- Идиопатический миелофиброз отличается образованием патологических элементов, и фиброзная ткань постепенно заменяет костный мозг.
- Истинная полицитемия густая кровь из-за обилия эритроцитов. Они накапливаются в селезенке и приводят к ее увеличению. При этом случаются кровотечения или тромбоз сосудов, что влечет за собой инсульт и инфаркт.
- Хронический миелолейкоз отличается большим скоплением лейкоцитов в костном мозге.
- Хронический нейтрофильный лейкоз характеризуется выработкой стволовыми клетками пораженных клеток-нейтрофилов (они призваны бороться со всевозможными инфекциями), из которых образуется опухоль. Развитие патологии идет медленно.
- Для эозинофильной лейкемии характерна чрезмерная выработка эозинофилов (вид лейкоцитов, который борется с инфекциями, вызванными некоторыми паразитами).
Миелопролиферативные заболевания часто переходят в острую форму лейкемии.
У таких заболеваний нет общепринятой системы стадирования, применяемой при обнаружении различных новообразований. Стратегию лечения выбирают в соответствии с видом патологии.
Существует 2 пути распространения опухоли в организме:
- Лимфогенный характеризуется проникновением патологии в органы по лимфатическим сосудам.
- Гематогенный отличается попаданием больных клеток в здоровые через кровеносную систему. Такой путь сопровождается образованием метастазов.
Симптоматика заболевания
Миелопролиферативный синдром каждого вида проявляется по-разному. Однако можно выделить некоторые общие симптомы:
- звон в ушах,
- быстрая и сильная утомляемость,
- кровотечения и кровоизлияния,
- отечность,
- болевые ощущения в суставах, левом предплечье и животе,
- быстрое снижение веса, часто приводящее к анорексии,
- селезенка или печень увеличиваются в размерах,
- лихорадочное состояние, сочетающееся с появлением синюшных пятен на лице и конечностях.

Для диагностирования проводят обследование пациента, включающее следующие процедуры:
- забор крови для анализа,
- исследование мазка,
- биопсия,
- полимеразная цепная реакция,
- исследование клеток для установления изменений в ph-хромосомах.
После постановки диагноза пациенту необходимо наблюдение у врача-гематолога.
Методы лечения
Лечение миелопролиферативных заболеваний крови проводится несколькими методами. Из числа самых эффективных можно выделить следующие:
- Химиотерапия характеризуется применением цитостатических препаратов, которые разрушают имеющиеся патогенные клетки, предотвращают появление других. Их вводят в кровь посредством инъекций или перорально. Этот способ называется системным. А также используют региональный метод лекарство вводят напрямую в спинномозговой канал или больной орган.
- Флеботомия заключается в снижении количества эритроцитов.
- Лучевая терапия основывается в использовании излучения высокой частоты, разрушающего новообразования, а также замедляющего появление опухолей.
- Аферез тромбоцитов основан на снижении уровня этих клеток посредством очищения крови через специальный сепаратор.
- Трансфузия это переливание крови с заменой одних компонентов на другие.
Миелопрофилеративные заболевания вылечить достаточно сложно и долго, поэтому при первых признаках необходимо обратиться к врачу.
алло-ТГСК - аллогенная трансплантация гемопоэтических стволовых клеток
АЧТВ - активированное частичное тромбопластиновое время
ВОЗ - Всемирная организация здравоохранения
ГЭС - гиперэозинофильный синдром
ИТК - ингибиторы тирозинкиназ
КТ - компьютерная томография
ЛПЗ - лимфопролиферативное заболевание
МО - молекулярный ответ
МПЗ-эо - миелопролиферативное заболевание с эозинофилией
МРТ - магнитно-резонансная томография
ПГО - полный гематологический ответ
ПЦО - полный цитогенетический ответ
ПЦР - полимеразная цепная реакция
РКИ - рандомизированные контролируемые исследования
СЦИ - стандартное цитогенетическое исследование
УЗИ – ультразвуковое исследование
ХМЛ - хронический миелолейкоз
CEL-NOS - chronic eosinophilic leukemia, not otherwise specified/хронический эозинофильный лейкоз, никак иначе не определяемый
FGFR1 - ген, кодирующий синтез рецептора к ростовому фактору, продуцируемому фибробластами
FISH - флуоресцентная гибридизация
JAK2 - ген, кодирующий синтез тирозинкиназы jak2
NCI CTCAE - шкала токсичности Национального института Рака Канады
PDGFRA - ген, кодирующий синтез α-цепи рецептора к ростовому фактору, продуцируемому тромбоцитами/мегакариоцитами
PDGFRВ - ген, кодирующий синтез β-цепи рецептора к ростовому фактору, продуцируемому тромбоцитами/мегакариоцитами
Ph ´ - филадельфийская хромосома
УДД - уровень достоверности доказательств
УУР – уровень убедительности рекомендаций
1. 2018 Клинические рекомендации "Миелопролиферативные заболевания (МПЗ), протекающие с эозинофилией" (Национальное гематологическое общество).
Определение
Миелопролиферативные заболевания, протекающие с эозинофилией (МПЗ-эо) – это группа опухолевых заболеваний миелоидной ткани, в основе которых лежат структурные нарушения ряда генов. Ключевыми являются гены, кодирующие синтез активных белков - тирозинкиназ: PDGFRA, PDGFRB, FGFR1, JAK2 . В редких случаях МПЗ-эо патологический клон содержит другие гены в составе различных хромосомных аберраций. До открытия патогенетической роли молекулярных нарушений данные заболевания рассматривались как гиперэозинофильный синдром (ГЭС), то есть, совокупность абсолютной эозинофилии, равной или превышающей 1,5х10 9 /л, и различных клинических симптомов, причина которого осталась неустановленной.
Терминология
Гиперэозинофильный синдром – состояние, при котором абсолютное число эозинофильных лейкоцитов в крови превышает 1,5х10 9 /л, имеются признаки поражения внутренних органов (сердце, легкие, желудочно-кишечный тракт, кожа и т.д.).
Миелопролиферативные заболевания, протекающие с эозинофилией – заболевания, при которых у пациентов с гиперэозинофильным синдромом выявлены молекулярные и цитогенетические маркеры, подтверждающие клональную (опухолевую) природу заболевания.
Эозинофилия - состояние, при котором абсолютное число эозинофильных лейкоцитов в крови превышает 0,6х10 9 /л.
Критерии качества
Уровень убедительности рекомендаций
Уровень достоверности доказательств
Выполнена консультация врача-паразитолога
Выполнен клинический анализ крови с подсчетом лейкоцитарной формулы и уровня тромбоцитов по мазку
Выполнена коагулограмма: протромбиновый индекс, АЧТВ, фибриноген
СМОТРЕТЬ ДРУГИЕ КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
Год утверждения 2017
Профессиональные ассоциации
- Национальное гематологическое общество
Оглавление
1. Краткая информация
Миелопролиферативные заболевания, протекающие с эозинофилией (МПЗ-эо) – группа опухолей миелоидной ткани, в основе которых лежат структурные нарушения ряда генов.
Ключевые гены, кодирующие синтез активных белков - тирозинкиназ:
Редко МПЗ-эо клон содержит мутации других генов в составе различных хромосомных аберраций.
Ранее заболевания рассматривались как идиопатический гиперэозинофильный синдром (ИГЭС) - совокупность абсолютной эозинофилии от 1,5х10 9 /л и различных клинических симптомов.
Поликлональный процесс, регулируемый эозинофилопоэтическими цитокинами, стимулирующими пролиферацию эозинофилов и их предшественников.
Заболевания и состояния, сопровождающиеся реактивной эозинофилией:
- Паразитозы: описторхоз, трихинеллез, токсокароз, эхинококкоз, филяриоз, аскаридоз, стронгилоидоз, шистосомоз;
- Хронические инфекции;
- ВИЧ-инфекция;
- Период восстановления после бактериальных инфекций.
- Атопические заболевания: бронхиальная астма, аллергический ринит, атопическая экзема, крапивница;
- Пищевая аллергия;
- Лекарственная аллергия.
- Острая и хроническая идиопатическая эозинофильная пневмония (болезнь Леффлера).
- Эозинофильный эзофагит первичный или вторичный;
- Гастроэнтерит первичный или вторичный, включая целиакию;
- Колит первичный или вторичный.
Другие причины аутоиммунного, воспалительного или токсического характера:
- Заболевания соединительной ткани (склеродермия, узелковый периартериит, СКВ);
- Синдром Черга-Страусс (эозинофильный васкулит);
- Эозинофильный фасциит;
- Болезнь Кимура;
- Саркоидоз;
- Хронический панкреатит;
- Синдром эозинофилии-миалгии.
- Солидные опухоли (особенно с метастазами в костный мозг).
- Лимфопролиферативные заболевания (лимфомы);
- Клональные Т-лимфоцитов с аберрантным иммунофенотипом (CD3- CD4 +) без лимфопролиферативного заболевания
Эндокринная недостаточность надпочечников (болезнь Аддисона).
ИГЭС - диагноз исключения и лишь констатирует факт эозинофилии и поражения органов.
Предполагается либо реактивная, либо миелопролиферативная природа синдрома.
В США заболеваемость миелопролиферативным вариантом ГЭС - 0,036 на 100 тысяч взрослых.
С92.7 – Другой миелоидный лейкоз;
D79.1 – Эозинофилия.
Нозологические формы (ВОЗ, 2008):
1. Миелоидные новообразования с аномалиями гена PDGFRA;
2. Миелоидные новообразования с аномалиями гена PDGFRВ;
3. Миелоидные новообразования с аномалиями гена FGFR1;
4. Хронический эозинофильный лейкоз, никак иначе не определяемый (CEL-NOS).
5. Гиперэозинофильный синдром – отличается от CEL-NOS числом бластных клеток.
2. Диагностика
- на анемические жалобы, симптомы гиперметаболического состояния (субфебрилитет, потеря веса);
- на сопутствующие заболевания и их терапии;
- на консультацию паразитолога с комплексным обследованием;
- по показаниям проведенные консультации специалистов (кардиолога, невропатолога, ревматолога и др.);
- оценка анамнеза и предшествующего обследования для исключения заболеваний, сопровождающихся реактивной эозинофилией.
- Осмотр с измерением роста и массы, температуры тела;
- Оценка костно-суставной системы;
- Выявление анемического и геморрагического синдрома;
- Наличие гепатоспленомегалии, лимфоаденопатии;
- Наличие дисфункции сердца, легких, печени, других органов.
1. Клинический анализ крови с лейкоцитарной формулой, тромбоцитами, ретикулоцитами;
2. БАК: общий билирубин, АСТ, АЛТ, ЛДГ, мочевая кислота, мочевина, креатинин, общий белок, альбумин, ЩФ, электролиты (K, NA, Ca, F, Mg), амилаза, липаза, глюкоза;
3. Коагулограмма: протромбиновый индекс, АЧТВ, фибриноген - для выявления гиперкоагуляции как осложнения эозинофилии;
4. Иммунохимическое исследование белков сыворотки с уровнем IgE;
5. Уровень сердечного тропонина – миокардит как осложнение эозинофилии.
Подозрение на миелопролиферативный процесс:
- миелоцитарный сдвиг в лейкоцитарной формуле
- анемия
- тромбоцитопения или тромбоцитоз
- нормальный уровень IgE.
Для точной верификации нозологической формы необходимы:
1. миелограмма;
2. трепанобиопсия;
3. СЦИ костного мозга, лимфатических узлов (при увеличении);
4. ПЦР с праймерами FIP1L1-PDGFRА, ETV6-PDGFRВ костного мозга, лимфоузлов (при увеличении);
5. FISH с зондами для выявления структурных нарушений генов PDGFRА, PDGFRВ, FGFR1 костного мозга, лимфоузлов (при увеличении);
6. При отрицательном PDGFRА - исключения других редких аберраций генов PDGFRВ, FGFR1 (FISH и/или ПЦР).
- УЗИ органов брюшной полости; средостения - для выявления увеличенных лимфоузлов;
- Эхо КГ;
- ЭКГ стандартная в 12 отведениях;
- Рентгенография грудной клетки;
- КТ грудной и брюшной полости, малого таза для исключения других опухолей и заболеваний;
- МРТ;
- Биопсия органов и патологических новообразований для верификации характера поражения;
- МРТ головного мозга - при наличии общемозговой и очаговой симптоматики.
- FISH и/или качественная ПЦР костного мозга или крови на химерный ген BCR-ABLпри характерных для ХМЛ клинико-лабораторных симптомах и неинформативности СЦИ.
Исключение системного мастоцитоза:
- в трепанобиоптате и/или других биоптатах (кроме кожи) множественные очаговые (≥ 15 в агрегате) скопления тучных клеток;
- в миелограмме >25% тучных клеток с атипичной формой (веретенообразные и др.);
- при ПЦР в биоптатах D816V-мутации гена с-KIT;
- при иммунофенотипировании обнаружение тучных клеток, экспрессирующих CD2 или CD25;
Для верификации лимфопролиферативных заболеваний (ЛПЗ) при характерных симптомах:
1. ИГХ биоптата ЛУ/удаленной селезенки;
2. Иммунофенотипирование;
3. ПЦР для выявления реаранжировка генов Т- и В-клеточных рецепторов – IgVH, TCR.
Перспектива трансплантации для пациентов:
- с аномалией гена FGFR1 при FISH;
- с агрессивным течением заболевания;
- с резистентностью к иматинибу у молодых больных.
Для поиска донора:
- при наличии сиблингов - HLA-типирование
- при отсутствии сиблингов - поиск HLA-совместимого неродственного донора.
3. Лечение
- максимальная редукция опухолевого клона;
- снижение риска прогрессии;
- предотвращения жизнеугрожающих специфических осложнений;
- нормализация состояния и повышение качества жизни.
При использовании ингибиторов тирозинкиназ (ИТК):
- эффективность терапии PDGFRA- и PDGFRВ-позитивных МПЗ-эо приближается к 100%;
- существенно улучшился прогноз – увеличилась продолжительность жизни, снизилась частота развития тяжелых специфических осложнений.
- 100 мг/сут при PDGFRA- позитивных.
- 400 мг/сут при PDGFRВ-позитивных.
- 400 мг/сут при CEL-NOS.
- 400 мг/сут при миелопролиферативном варианте ИГЭС.
Режим приема иматиниба:
- ежедневно
- длительно
- во время еды
- запивать полным стаканом воды.
Абсолютных противопоказаний для иматиниба нет, но с осторожностью:
- удлиненный QT,
- клинически выраженная сердечная недостаточность,
- дисфункция левого желудочка,
- аритмии.
Оценка эффективности иматиниба на основании мониторинга гематологических, цитогенетических и молекулярно-генетических показателей.
Для раннего выявления возможной токсичности показан регулярный мониторинг биохимических показателей крови, физикальный осмотр.
При PDGFRA-позитивном новообразовании в абсолютном большинстве:
- быстрый и полный гематологический ответ (ПГО);
- показатели крови и соматический статус нормализуются в течение первого месяца лечения;
- исчезновение транскрипта FIP1L1-PDGFRA на 2-4 месяце;
- для исключения рецидива после ПМО продолжать приём иматиниба.
При PDGFRA- и PDGFRВ-позитивных новообразованиях основное подтверждение эффективности терапии и почти 100% безрецидивная выживаемость:
- полный клинико-гематологический ответ
- цитогенетический/молекулярный ответ.
При CEL-NOS, миелопролиферативном варианте ИГЭС оценка эффективности по данным трепанобиопсии в совокупности с клинико-лабораторными изменениями.
Критерии ответа на терапию
Полный гематологический ответ:
- Лейкоциты менее 10.0х10^ 9 /л
- Эозинофилы менее 0,6х10^ 9 /л
- В гемограмме не повышен процент миелоцитов, промиелоцитов, миелобластов
- Тромбоциты более 150.0х10^ 9 /л
- Гемоглобин более 120 г/л
- Селезенка, печень не пальпируются
- Отсутствие всех симптомов и жалоб, обусловленных клональным новообразованием/ гиперэозинофилией
При варианте CEL-NOS, диагностированном по повышенным бластам (патологический клон не выявлен/кариотип не известен), дополнительно учитываются:
- Миелограмма - эозинофилы менее 10%, бласты менее 5%, клеточность не повышена
- Трепанобиоптат – нормальное соотношение жирового и деятельного костного мозга, клеточных линий миелопоэза
Первый признак эффективности иматиниба:
- нормализация числа эозинофилов периферической крови в первые 1 - 3 недели в большинстве случаев
Полный цитогенетический ответ - не определяются, обнаруженные в дебюте:
- перестройки генов PDGFRA, PDGFRB, FGFR1 при FISH
- хромосомные аномалии – методом СЦИ при анализе не менее 20 метафаз
Полный молекулярный ответ - не определяются, обнаруженные в дебюте молекулярные маркеры:
- FIP1L1-PDGFRA,
- ETV6-PDGFRB
- исчезают незрелые клетки лейкоцитов.
- отражают общую динамику ответа и проявляются до 4 месяцев терапии;
- в единичных случаях FIP1L1-PDGFRA-позитивного более позднее - после 12-го месяца, не влияющее на безрецидивную выживаемость.
Показание для повышения дозы иматиниба - эозинофилия свыше 1.0х10^ 9 /л более 3 недель, либо в любые сроки признаки прогрессирования:
- до 400 мг/сут при начальных 100 мг/сут;
- до 600 мг/сут при начальных 400 мг/сут.
Резистентность к препарату - отсутствие положительной динамики эозинофилии или ухудшение течения 2 недели после повышения дозы.
При резистентности к иматинибу:
Рекомендации по лечению специфических осложнений гиперэозинофилии
Адекватная циторедуктивная терапия – основной метод профилактики и лечения осложнений.
Лейкоцитаферез при необходимости быстрого уменьшения клеточной массы в циркуляции.
При гиперкоагуляционном синдроме с тромбоэмболическими осложнениями:
- гепарин 24 000МЕ/сут;
- низкомолекулярный гепарин (эноксапарин, фраксипарин, далтепарин).
Может потребоваться при развитии фибропластического эндокардита.
Первые доклинические признаки специфического поражения сердца выявляются при Эхо КГ.
- утолщение стенок желудочков,
- утолщение межжелудочковой перегородки,
- укорочение створок клапанов, очень часто – задней створки митрального клапана, с регургитацией,
- формирование фиброза и нарушение эластичности стенок,
- уменьшение объема желудочков – рестриктивная кардиопатия с тяжелой с НК, уменьшением сердечного выброса, застойными явлениями.
Хирургическая коррекция фибропластического эндокардита при достижении полной и стабильной ремиссии - протезирование створок клапанов.
Алло-ТГСК:
1. FGFR1-позитивное новообразование;
2. резистентность к иматинибу и другим вариантам консервативной терапии.
4. Реабилитация
Реабилитация при специфических осложнениях: поражение сердца, нервной системы.
Соответствующие патологии реабилитационные программы.
5. Профилактика
Пожизненное диспансерное наблюдение.
При полном гематологическом ответе либо стабильном течении заболевания наблюдение, в среднем, 1 раз в год.
6. Дополнительная информация, влияющая на течение и исход заболевания
Частота обследования больных, получающих иматиниб:
Клинический анализ крови:
- каждые 7 дней до достижения и подтверждения ПГО;
- каждые 3 месяца или по мере необходимости - при стабильном ответе.
Мониторинг обнаруженной в дебюте заболевания генетической аномалии:
- каждые 3 месяца до достижения и подтверждения ПЦО/ПМО;
- каждые 6 месяцев в первые два года после ПЦО/ПМО;
- один раз в год с 3 года ПЦО/ПМО.
Подсчет миелограммы, трепанобиопсия (только для CEL-NOS, миелопролиферативного ИГЭС):
- через 3 месяца от начала лечения;
- далее каждые 6 месяцев до достижения нормализации состояния костного мозга.
Биохимический анализ крови:
- 1 месяц терапии каждые 14 дней;
- первые 3 месяца терапии 1 раз в месяц,
- после 3 месяцев 1 раз в 3 месяца;
- для оценки токсичности показан более частый контроль.
Читайте также: