
Миелолейкоз гены bcr abl

Что такое хронический миелолейкоз?

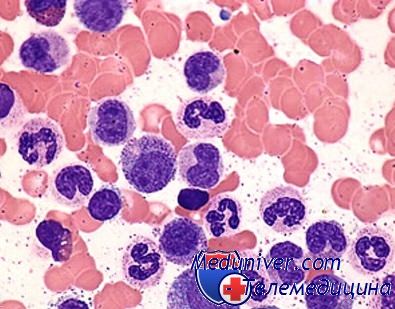
Мазок крови пациента с хроническим миелолейкозом
Хронический миелолейкоз (ХМЛ) — злокачественное новообразование кроветворной ткани, сопровождающееся прогрессирующей пролиферацией незрелых гранулоцитов. Заболевание изначально обладает вялотекущим характером, постепенно перетекая в стадию обострения с выраженной симптоматикой и образованием системных нарушений. Является одной из самых опасных и инвалидизирующих болезней.
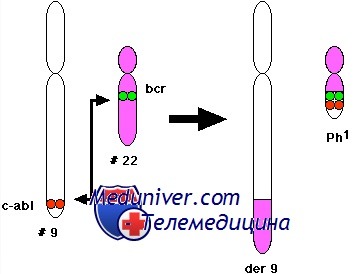
ХМЛ — первое онкологическое заболевание, у которого определена связь между развитием канцерогенеза и мутацией в гене. Характерная аномалия основана на транслокации 9-й и 22-й хромосом, то есть участки данных хромосом меняются местами, образуя аберрантную хромосому. Выявлена мутировавшая хромосома исследователями из Филадельфии, поэтому она получила название филадельфийская или Ph-хромосома.
Причины развития

Негативное воздействие на кроветворение оказывают ядохимикаты
Заболевание известно науке с 1811 года, но до сих пор факторы, провоцирующие мутацию в гене, определить не удалось. Существует ряд причин, способствующих развитию патологии:
- радиоактивное облучение, в том числе при лучевой терапии;
- химиотерапия иных онкологических заболеваний;
- ряд генетических заболеваний, характеризующихся хромосомной аномалией (например, синдром Дауна);
- взаимодействие с химическими соединениями (нефтепродукты, пестициды).
Патогенез хронического миелолейкоза

Патогенез хронического миелолейкоза
Гибридный ген BCR-ABL 1, образованный в результате транслокации хромосом, продуцирует синтез белка BCR-ABL. Данный белок представляет собой тирозинкиназу, которая в норме способствует передаче сигнальных импульсов для роста клетки. Созданная путём мутации тирозинкиназа становится активным фактором пролиферации клеток, они начинают делиться и распространяться уже независимо от факторов роста. Происходит процесс создания клонов мутировавшей клетки.
Бесконтрольное деление сопровождается нарушением апоптоза — запрограммированной гибели клеток. Также гибридная тирозинкиназа подавляет естественные функции восстановления в молекулах ДНК, создавая предпосылки для последующих мутаций, что усугубляет патологический процесс.
Размножающиеся клетки являются незрелыми, бластными предшественниками полноценных элементов крови. Постепенно бластные клетки вытесняют функциональные эритроциты, тромбоциты и лейкоциты. Добавляются нарушения и в других хромосомах, что запускает ускоренный процесс разрушения организма в целом.
Стадии хронического миелолейкоза

Бластный криз — одна из стадий миелолейкоза
- Хроническая — 30% бластных клеток. Стадия характеризуется агрессивным характером мутировавших клеток, состояние пациента резко ухудшается. Дополнительные аномалии как в гене BCR-ABL, так и в геноме в целом, провоцируют цепь патологических реакций, которые уже практически не поддаются лечению. На этом этапе могут поражаться ткани внутренних органов, кожные покровы и слизистые оболочки, миелоидные клетки преобразовываются в саркому.
Симптомы и признаки

Признаки ХМЛ становятся заметны ближе к прогрессирующей стадии.
- Симптомы опухолевой интоксикации: снижение массы тела, быстрая утомляемость, волнообразное повышение температуры, кожный зуд, тошнота, суставные боли.
- Симптомы опухолевой пролиферации — увеличение селезёнки и печени, боль в левом подреберье, поражение кожных покровов.
- Анемический синдром — головокружение, выраженная бледность, учащённое сердцебиение, чувство нехватки воздуха.
- Геморрагический синдром — склонность к кровоточивости слизистых оболочек, сыпь в виде красных точек, длительное кровотечение при незначительных порезах.
Диагностика заболевания

Один из методов диагностики заболевания — рентгенологический
Диагностика ХМЛ включает:
- Первичный осмотр пациента с изучением анамнеза, жалоб, а также исследование при помощи пальпации размеров селезёнки и печени.
- Общий анализ крови выявляет число и характеристики форменных элементов крови.
- Биохимический анализ проводится для определения уровня билирубина, электролитов, глюкозы, ЛДГ, АСТ, АЛТ.
- Гистологическое исследование костного мозга определяет скопления бластных клеток.
- Цитогенетический анализ выявляет транслокацию хромосом.
- На 3-й стадии проводится иммунофенотипирование для идентификации бластных клеток.
- Метод генного секвенирования применяется для выявления генных мутаций.
- Проводится УЗИ внутренних органов, в первую очередь селезёнки и печени.
- Дополнительно назначают рентгенографию органов грудной клетки, ЭКГ, эхокардиографию, ИФА на маркеры различных заболеваний, коагулограмму и другие исследования.
Лечение

Основа лечения — ингибиторы тирозинкиназы
Выбор препарата и доза определяются в зависимости от стадии ХМЛ и риска побочных эффектов. Обычно лечение начинается с приёма иматиниба в дозировке 400 мг/день при начальной стадии, 600 мг/день при последующих стадиях, затем дозу могут увеличивать или снижать. Различные аберрации в генах обусловливают низкую чувствительность к препаратам, поэтому пациенту могут менять одни ингибиторы на другие.
![]()
Трансплантация костного мозга
Если терапия не оказывает действия, рекомендуется аллогенная трансплантация костного мозга. Новые стволовые клетки могут выработать здоровые элементы кровеносной системы. Но операция сопряжена с рядом высоких рисков.
Терапия препаратами интерферона назначается обычно в 1-й стадии ХМЛ, так как не обладает эффективностью при последующих.
Для уменьшения массы опухоли и при отсутствии результата в лечении ингибиторами проводится химиотерапия. В стадии бластного криза используется полихимиотерапия аналогично лечению острого лейкоза.
Лучевая терапия может быть назначена в случае выраженной спленомегалии. При риске разрыва селезёнки проводят спленэктомию.
Профилактика и прогноз

Прогноз заболевания определяет врач
Причина образования ХМЛ не установлена, поэтому профилактикой являются меры по избеганию контактов с канцерогенными веществами, воздействия радиоактивного облучения.
Прогноз определяется стадией и тяжестью болезни. Одна из прогностических моделей (Kantarjian H.M.) включает факторы:
- преклонный возраст пациента при постановке диагноза;
- концентрация бластных клеток в крови ≥ 3%, в костном мозге ≥ 5%;
- концентрация базофилов ≥ 7%;
- концентрация тромбоцитов ≥ 700*10 9/л;
- выраженная спленомегалия.
Молекулярно-биологическое исследование, направленное на выявление точечных мутаций в гене BCR-ABL, которые могут обуславливать резистентность хронического миелолейкоза к некоторым ингибиторам тирозинкиназ.
Определение мутаций киназного домена BCR-ABL.
BCR/ABL1, Tyrosine Kinase Inhibitor Resistance, Kinase Domain Mutation Screen; Kinase domain mutation, ABL.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Специальной подготовки не требуется.
Общая информация об исследовании
Хронический миелолейкоз является опухолевым заболеванием кроветворной системы, в результате которого в периферической крови наблюдается повышение уровня определенного вида лейкоцитов. Развитие заболевания связано с возникновением в стволовой клетке крови генетической аномалии, которая получила название филадельфийской хромосомы. У человека на девятой хромосоме локализуется ген ABL, который кодирует образование белка, стимулирующего рост и деление клеток. При хроническом миелолейкозе часть гена ABL перемещается на хромосому 22, такая мутация называется транслокацией. Место разрыва 22-й хромосомы, куда чаще всего встраивается перемещенная часть гена ABL, называется M-bcr (от английского major breakpoint claster region). В норме в этом участке хромосомы расположен ген BCR, кодирующий белок, функция которого в настоящее время достоверно не изучена, однако есть сведения, что он участвует в процессах деления и дифференцировки клеток. В результате транслокации на 22-й хромосоме образуется сливной, или химерный, ген BCR-ABL, при считывании информации с которого продуцируется белок р210. От части гена ABL этот белок получает способности тирозинкиназы – внутриклеточного фермента, участвующего в передаче сигналов пролиферации, а часть, кодируемая геном BCR, способствует его активации. Таким образом, в результате описанной мутации на 22-й хромосоме формируется ген, белковый продукт которого – тирозинкиназа с повышенной активностью – стимулирует деление клеток с полной независимостью от внешних регуляторных механизмов. Лейкозные клетки становятся нечувствительными к сигналам самоуничтожения, приобретают способность выходить из костного мозга в периферическую кровь, не дожидаясь созревания.
В первое время лейкозные клетки полностью контролируются геном BCR-ABL, они сохраняют способность к дифференцировке и функционируют практически полноценно. Однако в процессе течения заболевания ген BCR-ABL индуцирует нестабильность генома, вследствие неконтролируемой пролиферации возникают точечные мутации, в том числе в самом гене BCR-ABL. Некоторые мутации могут приводить к перестройке той части молекулы BCR-ABL-тирозинкиназы, к которой присоединяются таргетные лекарственные препараты, подавляющие её активность. В результате этого препарат теряет способность связываться с мутантной тирозинкиназой и ингибировать её. По этой причине, несмотря на продолжающийся постоянный прием лекарства, отмечается прогрессирование заболевания. К настоящему времени обнаружено свыше 50 различных мутаций гена BCR-ABL, которые могут обуславливать резистентность опухолевого клона к одному или нескольким ингибиторам тирозинкиназ.
Для чего используется исследование?
- Для выявления точечных мутаций в химерном гене BCR-ABL, которые могут менять структуру кодируемого им белка и приводить к резистентности к отдельным препаратам группы ингибиторов тирозинкиназ.
Когда назначается исследование?
- У пациентов с хронической фазой хронического миелолейкоза:
- неудача лечения в установленные контрольные временные точки: недостижение большого цитогенетического ответа к 3 месяцам терапии, недостижение большого молекулярного ответа к 12 месяцам терапии;
- любые признаки утраты ответа (гематологический или цитогенетический рецидив);
- повышение уровня BCR-ABL на 1 логарифм (в 10 раз) и утрата большого молекулярного ответа.
- При прогрессировании заболевания в фазу акселерации или бластного криза.
Что означают результаты?
Результат исследования представляет собой название обнаруженной мутации гена BCR-ABL и сведения о её влиянии на чувствительность к определенным ингибиторам тирозинкиназ.
- Исследование позволяет обнаружить только известные на настоящий момент мутации. В связи с этим отрицательный результат не означает отсутствие редкой, неизученной мутации гена BCR-ABL и, соответственно, резистентности к терапии, особенно если на это указывают другие клинические и лабораторные данные.
- Мутация T315I обуславливает резистентность ко всем существующим ингибиторам тирозинкиназ, кроме понатиниба.
- Помимо точечных мутаций гена BCR-ABL, есть и другие механизмы формирования резистентности к ингибиторам тирозинкиназ: активация дополнительных сигнальных путей, которые в обход BCR-ABL-тирозинкиназы усиливают деление и подавляют гибель опухолевых клеток, избыточное связывание препаратов с транспортными белками крови (например, связывание иматиниба с α1-кислым гликопротеидом приводит к повышению концентрации лекарства в плазме и снижению его поступления в опухолевые клетки), повышенная экспрессия белка множественной лекарственной устойчивости PGP (он подобно насосу выкачивает из цитоплазмы клеток лекарственные препараты). Таким образом, при отрицательных результатах исследования на мутации в гене BCR-ABL лечащий врач может порекомендовать дополнительное обследование для исключения других причин неэффективности терапии.
- Значимость выявленной мутации для смены ингибитора тирозинкиназ определяется исключительно лечащим врачом.
Клинический анализ крови: общий анализ, лейкоцитарная формула, СОЭ (с обязательной микроскопией мазка крови)
Кто назначает исследование?
Гематолог, онколог, терапевт, врач общей практики.
Литература
- Wintrobe’s clinical hematology / editors, John P. Greer, Daniel A. Arber, Bertil Glader, Alan F. List, Robert T. Means Jr., Frixos Paraskevas, George M. Rodgers. – 13th edition. Lippincott Williams & Wilkins, 2014. Pages 1711-1712.
- 95 % случаев хронического миелолейкоза
- 20 - 50% случаев острого В-лимфобластного лейкоза взрослых
- Ежегодно регистрируется 1-2 заболевших на 100 тысяч населения
- Пик заболеваемости: 30 - 50 лет
- Госпитализация не требуется
- Стабильное состояние на протяжении 4-5 лет
- Не более 5% бластных клеток в костном мозге и переферической крови
- "Легкие" симптомы: утомляемость, потливость, потеря веса, аритмия
- 85 % пациентов к моменту постановки диагноза находятся в хронической фазе
- 80% дети
- 20% взрослые
Гематология: национальное руководство / под ред. О. А. Рукавицына. - М. : ГЭОТАР-Медиа, 2015. С. 381-398.
Этиология и встречаемость хронического миелоидного лейкоза (ХМЛ). Хронический миелоидный лейкоз (ХМЛ) (MIM №608232) — клональная экспансия трансформированных кроветворных клеток-предшественниц, при которой возрастает число циркулирующих миелоидных клеток. Трансформация клеток-предшественниц происходит за счет экспрессии онкогена BCR-ABL.
Хронический миелоидный лейкоз (ХМЛ) составляет до 15% всех случаев лейкоза у взрослых и встречается с частотой 1-2 на 100 000; скорректированная возрастная встречаемость более высокая среди мужчин, чем среди женщин (1,3-1,7 против 1,0).
Приблизительно 95% пациентов с хроническим миелоидным лейкозом (ХМЛ) имеют филадельфийскую хромосому; остальные — сложные варианты транслокаций. Протоонкоген Абельсона (ABL), кодирующий нерецепторную тирозинкиназу, находится в сегменте 9q34, а ген точечного разрыва кластерного региона (BCR), кодирующий фосфопротеин, — в 22qll.
До настоящего времени функции белков Abl и Bcr в норме окончательно не определены. Белок Abl довольно хорошо сохраняется в ходе эволюции многоклеточных. Он присутствует как в ядре, так и в цитоплазме и связан с внутренней цитоплазматической мембраной. Относительное количество белка Abl в этих частях клетки изменяется в различных типах клеток, а также в ответ на стимулы.
Abl участвует в клеточном цикле, ответе на стресс, передаче сигналов от интегринов и в нервном развитии. Функциональные области Bcr включают двойную спираль для полимеризации с другими белками, область треонин-серин-киназы, область обмена ГДФ-ГТФ, вовлеченную в регуляцию белков семейства Ras, и область активации ГТФ, участвующую в регуляции Rac и Rho ГТФаз.
Экспрессия Abl не приводит к трансформации клеток, вызываемой экспрессией химерного белка Bcr-Abl. У трансгенных мышей, вырабатывающих химерный Bcr-Abl с рождения, развивается острый лейкоз, а инфицирование здоровых мышей ретровирусами, экспрессирующими Bcr-Abl, вызывает ряд острых и хронических лейкозов, в зависимости от генетического фона.
В отличие от белка Abl, химерный белок Bcr-Abl имеет активность конститутивной тирозинкиназы и ограничен цитоплазмой, где энергично связывает микрофибриллы актина. Bcr-Abl фосфорилирует несколько цитоплазматических субстратов и тем самым активизирует сигнальные каскады, управляющие ростом, дифференцировкой и, возможно, адгезией кроветворных клеток.
Неправильная активизация этого сигнального пути приводит к неуправляемому распространению гемопоэтических стволовых клеток, выходу незрелых клеток из костного мозга, и, в конце концов, к хроническому миелоидному лейкозу (ХМЛ).
По мере развития хронического миелоидного лейкоза (ХМЛ) становится все более агрессивным. В ходе этой эволюции клетки опухоли у 50-80% пациентов приобретают дополнительные хромосомные изменения [трисомию 8,i(17q), или трисомию 19], вторую филадельфийскую хромосому. Кроме описанных цитогенетических изменений, при развитии хронического миелоидного лейкоза (ХМЛ) также часто мутируют гены-супрессоры опухолевого роста и протоонкогены.
Хронический миелоидный лейкоз (ХМЛ) — двух- или трехфазная болезнь. Начальный, или хронический этап характеризуется незаметно подкрадывающимся началом с постепенным развитием усталости, недомогания, потери массы тела и минимальным или умеренным увеличением селезенки. Со временем хронический миелоидный лейкоз (ХМЛ) обычно переходит в фазу акселерации и затем в бластный криз, хотя некоторые пациенты переходят непосредственно от хронической фазы в бластный криз.
Развитие хронического миелоидного лейкоза (ХМЛ) включает появление дополнительных хромосомных аномалий в клетках опухоли, прогрессирующего лейкоцитоза, анемии, тромбоцитоза или тромбоцитопении, всевозрастающую спленомегалию, лихорадку и костные нарушения. Властный криз — состояние острого лейкоза, бласты могут быть миелоидными, лимфоидными, эритроидными или недифференцированными. Фаза акселерации — промежуточная между хронической фазой и бластным кризом.
Приблизительно 85% больных диагностируют в хронической фазе. В зависимости от метода исследования, средний возраст постановки диагноза колеблется от 45 до 65 лет, хотя заболеванию подвержены лица любых возрастов. При отсутствии лечения показатель перехода из хронической фазы в бластный криз составляет приблизительно 5-10% в первые 2 года и затем 20% за год. Поскольку бластный криз быстро приводит к летальному исходу, развитие криза равнозначно смерти.
Особенности фенотипических проявлений хронического миелоидного лейкоза (ХМЛ):
• Возраст начала: от середины до конца зрелости
• Лейкоцитоз
• Спленомегалия
• Усталость и недомогание
Выяснение молекулярной основы хронического миелоидного лейкоза (ХМЛ) привело к разработке специфического ингибитора тирозинкиназы Bcr-Abl — иматиниба (гливек). Теперь это лекарство — основной препарат при лечении хронического миелоидного лейкоза (ХМЛ). Более 85% больных дают четкий цитогенетический ответ на терапию иматинибом, с исчезновением t(9;22) в клетках, получаемых при пункции костного мозга.
Цитогенетический ответ соответствует значительному уменьшению тяжести хронического миелоидного лейкоза (ХМЛ) до уровня ниже 10 9 -10 10 лейкемоидных клеток. Тем не менее только у некоторых пациентов (
Что такое BCR-ABL?
BCR-ABL — гибридный белок, продукт гибридного гена BCR-ABL1, формирующегося в результатереципрокной транслокации между хромосомами 9 и 22 (филадельфийская хромосома). BCR-ABL является конститутивно активной тирозинкиназой, ответственной за онкогенную трансформацию клеток (онкобелком). Постоянная активность этой тирозинкиназы делает клетку нечувствительной к воздействию факторов роста и вызывает её избыточную пролиферацию..
Белок BCR-ABL существует в трёх формах: p190, p210 и p230, в зависимости от места прерывания BCR-фрагмента.
BCR-ABL - мишень для нескольких специально разработанных ингибиторов, которые успешно применяются для лечения хронического миелолейкоза.
Ошибочное присоединение части хромосомы 9 (ген ABL) к хромосоме 22 (ген BCR) вызывает образование мутантной хромосомы ( её называют филадельфийской, в честь города, в котором она была обнаружена). Результат этого аномального процесса - новый ген, который называется BCR-ABL.
Новый ген вызывает синтез нового белка, приводящего к формированию злокачественных клеток крови в костном мозге.
BCR - ABL провоцирует:
Хронический миелолейкоз (ХМЛ)
Данная форма лейкоза характеризуется ускоренной и нерегулируемой пролиферацией преимущественно миелоидных клеток в костном мозге с их накоплением в крови.
У абсолютного большинства людей с этим диагнозом наблюдается "филадельфийская хромосома" (BCR-ABL). К факторам риска, способствующим появлению болезни, относятся воздействие бензола и высоких доз радиации.
Большинство пациентов узнают о своей болезни случайно, после получения результатов обычного анализа крови. При данном диагнозе отмечается резкое увеличение количества молодых лейкоцитов. В случае, когда результаты анализа крови вызывают подозрение на лейкоз, пациента направляют на биопсию костного мозга.
Продолжительность хронической фазы зависит от того, насколько рано было диагностировано заболевание, а также от успешности проведённого лечения. Несвоевременное выявление болезни ведет к возникновению ускоренной стадии и острой форме лейкоза.
Лечение ХМЛ: таргетная (целевая) терапия ингибиторами тирозинкиназ: иматиниб, нилотиниб, дазатиниб и др. Данная терапия значительно улучшила показатели выживаемости.
Острый B-лимфобластный лейкоз (ОЛЛ)
Вероятность возникновения ОЛЛ несколько повышена у людей, проходивших лечение с использованием облучения или определенных видов химиотерапии.
Самые неблагоприятные прогнозы и сложное лечение болезни отмечается именно при наличии у больного мутантной филадельфийской хромосомы (BCR-ABL). Без лечения ОЛЛ приводит к гибели в течение нескольких месяцев или даже недель.
Вероятность достижения ремиссии:
Точечная мутация T315I в BCR-ABL области может вызвать резистентность пациента к назначаемым препаратам.
Следовательно, перед началом лечения следует подтвердить или опровергнуть наличие данной мутации.
Хронический миелоидный лейкоз (ХМЛ) — клональное новообразование, развивающееся из стволовых кроветворных клеток. Это первая опухоль человека, при которой был обнаружен характерный хромосомный маркер. Открытие сделано в 1960 г. американскими исследователями Р. С. Nowell и D. A. Hungerford в Филадельфии, поэтому маркер был назван филадельфийской (Ph)-хромосомой. Именно с этой находки началась клиническая цитогенетика в онкологии.
Филадельфийская хромосома — укороченная хромосома из группы так называемых малых акроцентриков. В каждой нормальной женской клетке эта группа включает две пары хромосом — 21-ю и 22-ю, а в мужских клетках таких хромосом не 4, а 5, поскольку в эту группу, кроме 21-й и 22-й хромосомных пар, включена также Y-хромосома.
Ph-хромосома выявляется при обычной окраске (без G-бендинга) практически у всех больных хроническим миелоидным лейкозом (95—98 %). На дифференциально окрашенных (бендинг) хромосомах видно, что укорочена одна из хромосом 22-й пары.
Примерно у 90 % больных Ph-хромосома видна во всех анализируемых метафазах, у остальных пациентов обнаруживают как клетки с Ph-хромосомой, так и нормальные клетки, причем в отдельных случаях Ph-хромосома регистрируется в меньшинстве клеток костного мозга. Транслокация (9;22) наблюдается в миелоидных клетках, эритробластах, В-и Т-лимфоцитах, мегакариоцитах. Этот факт свидетельствует о том, что заболевание начинается с некоммитированной клетки-предшественницы гемопоэза.
Примерно в 10 % случаев наблюдаются атипичные транслокации, при которых стандартное цитогенетическое исследование позволяет увидеть перенос фрагмента хромосомы 22 не на 9-ю, а на какую-либо другую хромосому. Кроме того, иногда при хроническом миелоидном лейкозе обнаруживают сложные Ph-транслокации с участием не двух (9-й и 22-й), а трех или более хромосом.
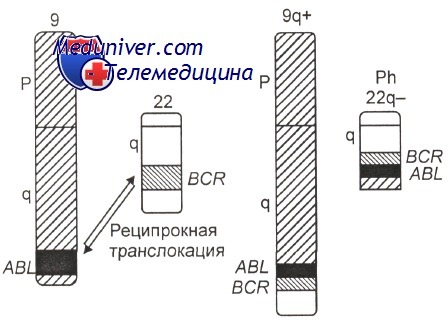
Возникновение химерных генов BCR-ABL и ABL-BCR в результате специфической хромосомной перестройки t(9;22)
Установлено, что практически при всех Ph-транслокациях участвуют хромосомы 9 и 22, однако это не всегда видно при стандартном цитогенетическом исследовании, но обнаруживается при использовании FISH и ПЦР.
Большинство исследователей считают, что тип Ph-транслокации (стандартная, атипичная или сложная) не имеет клинического значения.
Использование молекулярно-генетических методов позволило установить, что в хромосоме 9 разрыв проходит через ген (протоонкоген) ABL, идентифицированный ранее в одном из вирусов лейкоза у мышей. В хромосоме 22 наблюдается разрыв гена ВСЯ. В результате слияния фрагментов названных генов (ABL и BCR) образуется химерный ген BCR-ABL, расположенный, как правило, на делетированной хромосоме 22 (Ph-хромосома) Схематическое изображение молекулярно-генетических событий, приводящих к развитию хронического миелоидного лейкоза, показано на рисунке.
Примерно в 70 % случаев, кроме транскрипта BCR-ABL, обнаруживают продукт (транскрипт) другого химерного гена, образующегося в результате t(9;22) —ABL-BCR на der (9), однако его роль в развитии хронического миелолейкоза неясна.
Установлено, что делеция возникает одновременно с формированием специфической t(9;22), ее частота одинакова в группах больных, обследованных на разных стадиях хронического миелолейкоза. Независимо от метода лечения (даже при использовании программ, включающих гливек) прогноз у больных с интерстициальной делецией в маркере 9q+ существенно хуже, чем у больных без этой аномалии. Длительность хронической фазы и соответственно выживаемость у больных с делецией значительно меньше. Так, при обследовании большой группы больных (241) обнаружено, что медиана выживаемости была 38 мес для больных с делецией (39 человек) и 88 мес для группы больных без делеции (202 человека). Различия статистически значимы.
Применение этого информативного прогностического метода пока, к сожалению, не вошло в широкую клиническую практику, поскольку он весьма сложен и требует дорогостоящих реактивов и оборудования.
Роль делеции маркера 9q+ в прогрессии хронического миелолейкоза пока до конца не выяснена. Выпадение кодирующих последовательностей генов ABL и/или BCR в результате делеции приводит к тому, что экспрессируется только один химерный ген BCR-ABL, но нет экспрессии гена ABL-BCR. Возможно, и это событие играет роль в профессии лейкоза. Обсуждается также возможность инактивации неизвестных пока генов-супрессоров, локализованных в хромосомном районе, который делетируется.
Химерный ген BCR-ABL кодирует белок с мол. м. 210 000, обладающий более высокой протеинкиназной активностью, чем продукт нормального протоонкогена ABL (Р145). При лейкозе, вызванном у мышей вирусом Абельсона, онкогенной активностью обладает белок — продукт гибридного гена gag/abl с высокой протеинкиназной активностью. В эксперименте проводили вырезание гена gag/abl, после этого вирус терял способность вызывать лейкоз у мышей.
Изучение разрывов в генах ABL и BCR при хроническом миелолейкозе показало, что у разных больных их локализация неодинакова. Так, в гене ABL протяженность участка, на котором могут происходить разрывы, велика —до 200 kb, а в гене BCR разрывы локализуются обычно на маленьком участке — 8,5 kb, т. е. имеется кластер разрывов, отсюда название самого гена BCR (breakpoint cluster region).
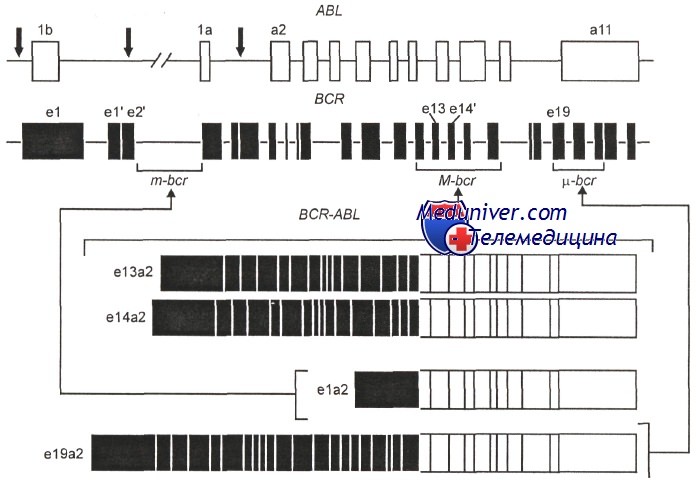
Разрывы в генах ABL и BCR при хроническом миоелоидном лейкозе.
Первый ряд — разрывы в гене ABL; второй ряд — три кластера разрывов в гене BCR: m-BCR, М-BCR и u-BCR. Ниже три типа транскриптов химерных генов BCR-ABL, различающихся по длине вошедших участков гена BCR
В подавляющем большинстве случаев t(9;22) разрывы гена BCR обнаруживают на участке, обозначаемом M-BCR, при этом химерный ген включает длинный фрагмент гена BCR и в результате возникает характерный для хронического миелолейкоза белок P210Bcr/Abl. Кроме того, при t(9;22) разрывы в гене BCR могут локализоваться на одном из двух других типичных участков, называемых m-bcr и u-bcr. Разрывы в области m-bcr приводят к образованию химерного белка Р190Bcr/Abl, т. е. белка меньшей величины, чем Р210Bcr/Abl. При локализации разрывов в u-bcr образуется более крупный белок Р230Bcr/Abl.
Как отмечалось, коррелятивная связь между типом химерного белка (P190Bcr/Abl, Р210Bcr/Abl или Р230Bcr/Abl) и клинико-гематологичесой картиной лейкоза не является строгой. Любой из этих белков может быть обнаружен при классической картине хронического миелолейкоза. Есть также сообщения о нередком сочетании двух типов белков (Р210Bcr/Abl| и Р190Bcr/Abl) при типичном хроническом миелолейкозе и при остром лимфобластном лейкозе.
Решающая роль гена BCR-ABL и его продукта — белка Р210 в развитии хронического миелолейкоза продемонстрирована на разных модельных системах in vivo и in vitro. Так, трансдукция bcr/abl в стволовые гемопоэтические клетки мыши с последующей трансплантацией этих клеток облученным сингенным животным вызывает у них миелопролиферативное заболевание, сходное с хроническим миелолейкозом человека. Установлено, что онкогенный потенциал химерного белка BCR-ABL обусловлен его высокой тирозинкиназной активностью. Дерегуляция тирозинкиназной активности — одно из центральных событий в злокачественной трансформации клеток.
При введении клеток, экспрессирующих р210Bcr/Abl, группе летально облученных мышей в условиях одного и того же эксперимента у одних животных развивался лейкоз, очень похожий на хронический миелолейкоз человека, а у других — самые разнообразные новообразования из гемопоэтических клеток: миеломоноцитарные лейкозы, макрофагальные опухоли, пре-В- и Т-клеточные лимфомы, ретикулоклеточные саркомы и эритроидные опухоли. Причина различий не выяснена. Эти опыты, как и эксперименты с трансгенными мышами, показывают, что вся цепь событий, приводящая к развитию картины хронического миелолейкоза, пока еще не установлена.
Получены данные, свидетельствующие о том, что масса кроветворных элементов и клеток крови в организме больных хроническим миелолейкозом существенно увеличена, главным образом за счет резкого повышения времени жизни этих клеток, поскольку активированный ген ABL (в гене BCR-ABL) ингибирует апоптоз — запрограммированную клеточную смерть. Кроме того, этот ген усиливает пролиферацию миелоидных клеток. Есть основания считать, что при хроническом миелолейкозе изменена функция специальных клеточных белков — интегринов, в результате чего нарушается адгезия молодых миелоидных клеток к стромальным элементам, и стволовые лейкемические клетки избегают негативных регуляторных влияний.
Читайте также: