
Хронический лимфоцитарный лейкоз клинические рекомендации
АГ антиген
АИГА аутоиммунная гемолитическая анемия
АТ антитело
БПВ беспрогрессивная выживаемость
БРВ безрецидивная выживаемость
БСВ бессобытийная выживаемость
ИТП идиопатическая тробоцитопеническая пурпура
КИК кумулятивный индекс коморбидности
МОБ минимальная остаточная болезнь
нПР нодулярная частичная ремиссия
ОВ общая выживаемость
ОО общий ответ
ПК периферическая кровь
ПР полная ремиссия
Пр прогрессия
Сб стабилизация
СКФ скорость клубочковой фильтрации
ХЛЛ хронический лимфолейкоз
ЧР частичная ремиссия
BR химиотерапевтический режим, содержащий бендамустин и ритуксимаб
CD кластер дифференцировки (cluster of differentiation)
CMV цитомегаловирус (citomegalovirus)
FC химиотерапевтический режим, содержащий флударабин и циклофосфан
FCR химиотерапевтический режим, содержащий флударабин, циклофосфан и ритуксимаб
FCMR химиотерапевтический режим, содержащий флударабин, циклофосфан, митоксантрон и ритуксимаб
FISH флуоресцентная гибридизация in situ
1. 2014 Клинические рекомендации по обследованию и лечению больных хроническим лимфолейкозом (Национальное гематологическое общество).
Критерии для постановки диагноза
Лимфоцитарная лимфома/хронический лимфолейкоз – это В-клеточная опухоль из мелких лимфоидных клеток с примесью пролимфоцитов и параиммунобластов. Компактное расположение пролимфоцитов и параиммунобластов образует псевдофолликулярные структуры.
Лимфоцитарная лимфома/хронический лимфолейкоз характеризуется гетерогенной экспрессией CD20, коэкспрессией CD5, CD23, CD43. Результаты иммуногистохимического окрашивания с антителами к CD38 и ZAP-70 не коррелируют с прогнозом.
Главным цитогенетическим маркером, непосредственно влияющим на выбор терапии, является делеция 17p (Ia). Рекомендуется проводить скрининг на делецию 17p у всех больных, имеющих показания к началу терапии. Hе рекомендуется проводить исследование FISH пациентам, у которых нет показаний к началу терапии. Кроме того, желательно исследовать наличие t(11;14), t(11q;v), +12, del(11q), del(13q).
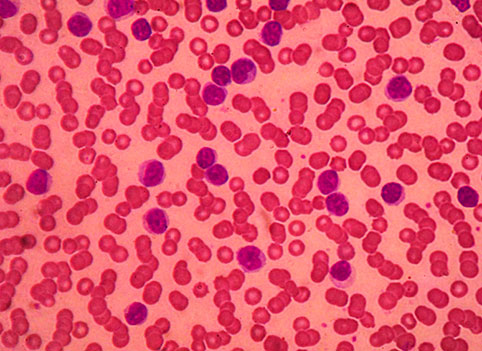
Для постановки диагноза хронического лимфолейкоза требуется анализ крови и иммунофенотипическое исследование. Диагноз устанавливается при выявлении более 5000 клональных В-лимфоцитов в 1 мкл периферической крови. Клетки ХЛЛ экспрессируют антиген CD5 и В-клеточные маркеры CD19, CD20, и CD23. Уровень экспрессии поверхностных иммуноглобулинов, CD20 и CD79b на клетках ХЛЛ ниже, чем на нормальных В-лимфоцитах. Для лимфомы из клеток мантийной зоны также характерна экспрессия CD5, но не характерна экспрессия CD23.В-клеточный пролимфоцитарный лейкоз характеризуется высокой экспрессией CD20; на клетках В-ПЛЛ в 50% случаев отсутствует CD5. В мазке крови клетки ХЛЛ выглядят как лимфоциты с узким ободком цитоплазмы и плотным ядром и частично агрегированным хроматином, без отчетливого ядрышка. Возможна примесь более крупных атипичных клеток, клеток с расщепленными ядрами и пролимфоцитов, это не противоречит диагнозу ХЛЛ. При выявлении более 55% пролимфоцитов в крови диагностируется В-клеточный пролимфоцитарный лейкоз. Для ХЛЛ характерно выявление в крови разрушенных клеток – теней Гумпрехта.
Лимфому из малых лимфоцитов диагностируют при наличии лимфаденопатии, спленомегалии, цитопений при условии, что число В-лимфоцитов в крови не превышает 5 х 109/л. Диагноз должен быть подтвержден биопсией лимфатического узла. В гистологических препаратах морфологический субстрат представлен диффузным ростом небольших лимфоидных клеток с округлыми ядрами, комковатым хроматином, без отчетливых ядрышек. В срезах ткани лимфатического узла нередко присутствуют фолликулоподобные структуры – псевдофолликулы, так называемые зоны роста, представленные увеличенным количеством параиммунобластов, клеток с морфологией пролимфоцитов с различимыми ядрышками. Псевдофолликулы могут демонстрировать слабую ядерную экспрессию Cyclin D1.
ХЛЛ или лимфому из малых лимфоцитов можно заподозрить у пациента с небольшим лимфоцитозом, но числом В-лимфоцитов
Схема 1. Критерии диагноза ХЛЛ, МBКЛ, ЛМЛ
У 3-5% больных ХЛЛ и лимфомой из малых лимфоцитов (аналога ХЛЛ) развивается крупноклеточная лимфома или лимфома Ходжкина. Появление крупноклеточной лимфомы на фоне ХЛЛ называется синдромом Рихтера. Появление лимфомы Ходжкина на фоне ХЛЛ называется ходжкинской трансформацией. Во всех случаях локального увеличения лимфоузлов или существенного изменения клинической картины болезни необходимо делать биопсию лимфоузла или экстранодального очага. Без морфологической верификации этот диагноз не ставится.
Определение стадии
Стадирование осуществляется по результатам обследования в соответствии с критериями классификации J.L. Binet (табл. 1)
Таблица 1. Стадии ХЛЛ по Binet
| Стадия | Характеристика | Медиана выживаемости, мес. | % больных в дебюте |
| A | Hb >100 г/л, тромбоциты >100 х 109/л Поражено | > 120 | 60% |
| B | Hb >100 г/л, тромбоциты >100 х 109/л Поражено >3 лимфатических областей | 61 | 30% |
| C | Гемоглобин | 32 | 10% |
Под лимфатическими областями понимают:
- шейные лимфоузлы
- подмышечные лимфоузлы (c одной или двух сторон)
- паховые лимфоузлы (c одной или двух сторон)
- печень
- селезенка
Алгоритм диагностики
Перед началом терапии (первой и последующих линий) рекомендуется выполнение следующих исследований:
- Осмотр с пальпацией лимфоузлов, печени, селезенки, осмотром миндалин
- Общий анализ крови c определением уровня тромбоцитов и ретикулоцитов
- Иммунофенотипирование крови, если не выполнялось ранее*
- Трепанобиопсия и миелограмма
- Биохимический анализ крови, включающий определение белка, билирубина, мочевой кислоты, трансаминаз и ЛДГ.
- Электрофорез и иммунохимическое исследование сыворотки крови и мочи
- Прямая проба Кумбса
- УЗИ брюшной полости. Ультразвуковое исследование периферических лимфоузлов относится к желательным, но не обязательным процедурам.
- Рентгенография грудной клетки
- Определение маркеров вирусов гепатитов В**, С и цитомегаловируса.
- Цитогенетическое исследование и/или FISH***
Остальные исследования выполняются исходя из клинических показаний. При подозрении на трансформацию показана биопсия лимфоузла, костного мозга, экстранодального очага.
*Минимальная панель иммунофенотипирования должна включать: CD19, CD5, CD23, CD79b, каппа и лямбда, FMC7, CD20, CD10, CD38, ZAP70. ** Всем больным, которым планируется назначение ритуксимаб-содержащих режимов, необходимо проводить развернутое вирусологическое обследование для выявления маркеров вируса гепатита B,
включающее Hbs-антиген и антитела к вирусу гепатита B (anti-Hbs, anti-Hbe). При выявлении антител к вирусу гепатита B (anti-Hbs) показано исследование ДНК вируса гепатита B в сыворотке. Наличие Hbs- антигена или наличие ДНК вируса гепатита у серопозитивного пациента является относительным противопоказанием к проведению терапии ритуксимабом. Для пациентов с гепатитом B лечение ритуксимаб-содержащими режимами должно проводиться при профилактическом назначении энтекавира. Энтекавир назначается на весь период лечения и не менее года после завершения терапии при неоднократных отрицательных результатах тестирования ДНК вируса гепатита B в крови. Оптимапльная продолжительность терапии энтекавиром после завершения терапии ритуксимабом не определена. ***Главным цитогенетическим маркером, непосредственно влияющим на выбор терапии, является делеция 17p. Рекомендуется проводить скрининг на делецию 17p у всех больных, имеющих показания к началу терапии. Hе рекомендуется проводить исследование FISH пациентам, у которых нет показаний к началу терапии. Кроме того, желательно исследовать наличие t(11;14), t(11q;v), +12, del(11q), del(13q).
Заболеваемость
Хронический B-клеточный лимфоцитарный лейкоз (В-ХЛЛ) наиболее частый вид лейкоза среди населения западного полушария, показатель заболеваемости составляет 4 случая на 100 тыс. чел. в год. Заболеваемость возрастает почти до 30 случаев на 100 тыс. чел. в год в возрасте > 80 лет. Медиана возраста на момент диагностики составляет 69 лет; 14% пациентов моложе 55 лет.
Диагноз
Диагноз В-ХЛЛ может быть поставлен на основании следующих критериев: диагноз В-ХЛЛ требует наличия . 5000 B- лимфоцитов/мкл периферической крови на протяжении минимум 3-х месяцев. Клональность циркулирующих лимфоцитов должна быть подтверждена методом проточной цитофлоуриметрии.
Клетки В-ХЛЛ, определяемые в мазках периферической крови имеют облик малых лимфоцитов ― ядра содержат плотный глыбчатый хроматин, ядрышко не визуализируется, цитоплазма представлена узким ободком.
Клетки В-ХЛЛ коэкспрессируют Т-клеточный антиген CD5 и поверхностные В-клеточные антигены CD19, CD20 и CD23. Уровень экспрессии поверхностных иммуноглобулинов, CD20 и CD79b лимфомными клетками ниже, по сравнению с нормальными В-лимфоцитами. Каждый лейкемический клон отличается экспрессией только одного вида легких цепей иммуноглобулинов- каппы или лямбды.
Для сравнения, клетки лимфомы зоны мантии, отличаясь также коэкспрессией CD5 и В-клеточных поверхностных антигенов, как правило не несут молекулу CD23. Дифференциальный диагноз должен быть также проведен с лимфомой маргинальной зоны и иммуноцитомой.
В соответствии с определением, лимфома из малых лимфоцитов (ЛМЛ) характеризуется наличием лимфаденопатии и/или спленомегалии. Количество лимфоцитов в периферической крои не должно превышать 5х109/л. ЛМЛ имеют иммунофенотип, идентичный В-ХЛЛ. Диагноз ЛМЛ должен быть подтвержден морфологическим исследованием биопсии лимфоузла.
План обследования пациента перед началом терапии должен содержать следующие обязательные пункты [III, B]:
- анамнез и тщательный осмотр, включающий пальпацию всех групп периферических л/у;
- развернутый анализ крови с подсчетом формулы;
- биохимический анализ крови, включая уровень ЛДГ, билирубина и иммуноглобулинов сыворотки;
- прямой антиглобулиновый тест;
- определение инфекционного статуса, включая гепатиты В, С, цитомегалови рус и ВИЧ;
- рентген грудной клетки;
- УЗИ органов брюшной полости
Проведение пунктов, представленных ниже, является желательным перед началом специфического лечения [III, B]:
- Биопсия костного мозга не требуется для подтверждения диагноза. Биопсия КМ выполняется перед началом миелосупрессивной терапии или при возникновении цитопении неясного генеза;
- Определение цитогенетических аномалий, в особенности делеции короткого плеча хромосомы 17 (del 17p) посредством флуоресцентной in situ гибридизации (FISH) имеет значение для выбора терапии, поэтому исследование рекомендуется к проведению перед началом лечения.
- Проведение КТ-исследования рекомендуется для оценки эффекта при проведении клинических исследований [III,C], но не для рутинной практики за рамками протоколов.
Стадирование и прогноз
Медиана выживаемости от момента диагностирования варьирует от года до более 10 лет в зависимости от начальной стадии болезни. В настоящее время используются две системы клинического стадирования. В Европе в основном используются система стадирования по Binet, на основании которой выделяются 3 разные прогностические группы (таблица №1).
C появлением новых терапевтических возможностей, наметилось улучшение показателей общей выживаемости пациентов с продвинутыми стадиями В-ХЛЛ.
В настоящее время выделены факторы, позволяющие определять прогноз у пациентов уже на начальных стадиях. В-ХЛЛ с del(17p) (5-10% всех случаев) имеет наихудший прогноз и медиану выживаемости 2-3 года. Другой фактор плохого прогноза — del (11q), которая определяется примерно в 20% случаев. Негативное влияние del (11q) на прогноз может, однако, быть преодолено иммунохимиотерапией с применением флударабина, циклофосфамида и ритуксимаба (FCR) (см. ниже).
Мутация IGHV (генов, кодирующих вариабельные участки тяжелых цепей иммуноглобулинов) отсутствует примерно в половине случаев В-ХЛЛ. Эти пациенты отличаются достоверно менее продолжительной общей выживаемостью и коротким временем до начала терапии.
Экспрессия CD38 и ZAP70 коррелирует в определенной степени с мутационным статусом IGHV. В отличие от молекулярных аномалий, определяемых FISH, данные факторы, однако, не должны влиять на выбор терапии, поскольку их значение еще должно быть установлено в клинических исследованиях [III, C].
Лечение начальных стадий
К начальным относятся бессимптомные А и В стадии по Binet и 0, I и II стадии по Rai.
Лечение должно быть начато при появлении признаков прогрессирования/активирования болезни, указанных ниже.
Лечение распространенных стадий заболевания (стадия по Binet А и В с симптомами активного процесса, Binet стадия С; стадия по Rai 0II с симптомами, Rai стадии IIIIV)
Лечению подлежат пациенты при наличии следующих признаков активации болезни: выраженные В-симптомы, цитопения, не являющаяся следствием аутоиммунных нарушений; осложнения, вызванные увеличением лимфатических узлов и сплено/гепатомегалией, а также больные с аутоиммунными анемией и тромбоцитопенией, плохо поддающимися терапии стероидами [I,A].
Соматическая сохранность и отсутствие тяжелой сопутствующей патологии должно лежать в основе выбора терапии.
Для начальной терапии соматически сохранных больных (физически активны, без серьезной сопутствующей патологии, с сохранной почечной функцией) терапией выбора служит схема FCR. Данный режим считается стандартом первой линии, поскольку, как было показано недавно, существенно влияет на улучшение показателей выживаемости [II,A]. Для соматически отягощенных пациентов, терапия хлорамбуцилом в первой линии остается стандартом лечения [II, B]. Альтернативой могут служить режимы на основании пуриновых аналогов в редуцированных дозах [III, B] или бендамустин [II, B].
Пациенты носители делеции 17р часто оказываются резистентны к стандартным режимам химиотерапии (монотерапии флударабином или комбинации FC). Введение в практику химиоиммунотерапии (FCR), также не повлияло на показатель безрецидивной выживаемости, которая остается непродолжительной. Таким образом, при наличии del(17p) должна быть предложена трансплантация аллогенных стволовых клеток в первой линии в рамках протоколов клинических исследований [III, B].
Вторая линия химиотерапии
Первая линия терапии может быть проведена повторно, если рецидив или прогрессирование развились более чем через 12 месяцев от начального лечения или спустя 24 месяца после иммунохимиотерапии [III, B].
Если рецидив развился в течение 12 месяцев от начального лечения или в течении 24-х месяцев после иммунохимиотерапии, а также при отсутствии эффекта от терапии первой линии, рекомендовано использование следующих препаратов и/или комбинированных схем:
- Алемтузумаб-содержащие режимы с последующей аллогенной трансплантацией для соматически сохранных пациентов
- FCR для пациентов резистентных или в рецидиве после терапии первой линии
- Алемтузумаб или бендармустин–содержащие режимы для соматически сохранных пациентов при отсутствии del(17p). В данной группе также возможна попытка применения офатумомаба или ритуксимаба в высоких дозах в сочетании со стероидами
- Алемтузумаб для пациентов- носителей del(17p).
Для достижения лучшего эффекта у пациентов с большой опухолевой массой, алемтузумаб может быть комбинирован с флударабином или стероидами.
Аллогенная трансплантация стволовых клеток является единственным методом излечения больных группы высокого риска (наличие del (17p) или del (11q)), а также во всех случаях рефрактерного течения болезни.
Аутологичная трансплантация стволовых клеток не показала преимущества в сравнении с современной иммунохимиотерапией и не должна более рассматриваться как терапевтическая опция при В-ХЛЛ [III, B].
Примерно в 10% случаев (3-16%) В-ХЛЛ развивается Синдром Рихтера, который представляет собой трансформацию в крупноклеточную лимфому, Лимфому Ходжкина или пролимфоцитарную лейкоз (ПЛЛ)***. Прогноз при Синдроме Рихтера, также как и при В-ПЛЛ очень плохой. Полихимиотерапия в сочетании с моноклональными антителами может быть предметом выбора, однако лечение, как правило, не приводит к получению длительных ремиссий.
Аллогенная трансплантация представляет собой экспериментальный подход, который, однако, может быть использован у соматически сохранных пациентов с Синдромом Рихтера.
***Синдром Рихтера представляет собой исключительно трансформацию В-ХЛЛ в диффузную В-клеточную крупноклеточную лимфому (прим. переводчика).
Оценка эффекта
Оценка эффекта лечения основана на контроле клинического анализа крови и тщательного осмотра пациента.
Исследование костного мозга показано только при достижении полной гематологической ремиссии.
Проведение рентгенологического исследования органов грудной клетки, ультразвукового исследования или компьютерной томографии органов брюшной полости для оценки эффекта проведенной терапии показано только в случае наличия регистрации изменений теми же методами перед началом лечения [V,D]. Определение минимальной резидуальной болезни (MRD) при помощи 4-х цветной проточной цитофлоуриметрии имеет прогностическое значение для длительности ремиссии. Пациенты с негативной MRD по окончании терапии обладают достоверно более длительной продолжительностью ответа. Клиническое значение негативной MRD однако до конца не ясно, поэтому анализ MRD имеет значение только в рамках клинических исследований, но не в рутинной практике.
Наблюдение
Наблюдение пациентов при отсутствии симптомов заболевания должно включать клинический анализ крови каждые 3-6 месяцев и тщательный регулярный осмотр лимфоузлов, печени и селезенки.
Особое внимание следует уделять появлению аутоиммунных цитопений (аутоиммунная гемолитическая анемия, аутоиммунная тромбоцитопения), встречающихся у 10-15% больных В-ХЛЛ.

Хронический лимфолейкоз (ХЛЛ)/лимфома из малых лимфоцитов – лимфопролиферативное заболевание, морфологическим субстратом которого являются опухолевые клональные лимфоидные клетки, имеющие размеры и морфологию зрелого лимфоцита и иммунофенотип, соответствующий В-лимфоцитам поздних стадий дифференцировки.
В странах Европы и Северной Америки ХЛЛ наиболее распространённый вид лейкоза. В этих странах на его долю приходится около 30 % от всех лейкозов. Ежегодная заболеваемость составляет 3–3,5 на 100000 населения, увеличиваясь до 20 на 100000 после 70 лет. Крайне редок ХЛЛ среди узбеков. Среди жителей Японии ежегодно регистрируется не более одного нового случая в год [2].
Чаще ХЛЛ встречается у мужчин, соотношение заболевших мужчин и женщин составляет 2:1, средний возраст 72 года. Почти 70 % больных – старше 65 лет, большинство из них к этому времени имеют несколько сопутствующих заболеваний. Это особенно важно учитывать, когда, после химиотерапии, развиваются многочисленные осложнения, обостряются хронические заболевания. Медиана возраста больных, умерших от ХЛЛ, составляет 79 лет [23]. Инфекционные осложнения являются основной причиной смерти больных с ХЛЛ. Смертность от инфекций составляет 30–50 % от всех случаев с летальным исходом [31].
Несмотря на существенный прогресс в терапии, ХЛЛ остаётся неизлечимым заболеванием. Целью лечения в настоящее время является увеличение доли общей выживаемости (ОВ) и выживаемости без прогрессирования (ВБП) при минимальном уровне токсичности. Это особенно важно у пожилых людей, имеющих сопутствующие заболевания [40].
ХЛЛ - самая частая форма лейкоза у кровных родственников [42]. Средний возраст на момент диагностики заболевания среди семейных случаев составляет 58 лет [26].
При исследовании течения болезни у 16367 больных ХЛЛ с 1973 по 1996 г. выявлено увеличение частоты вторых опухолей в 1,2 раза [25]. Установлен более высокий риск развития меланомы, лимфомы Ходжкина и острых миелоидных лейкозов среди больных, получавших лечение алкилирующими препаратами, и не был повышен среди не леченных больных и леченных только флударабином [36].
Критериями диагноза ХЛЛ является обнаружение в периферической крови В-лимфоцитов в количестве, превышающем 5 × 10 9 /л, наличие в аспирате костного мозга не менее 30 % лимфоцитов и обнаружение специфических маркёров методом проточной цитометрии. Клетки ХЛЛ коэкспрессируют антиген СD5 и В-клеточные маркёры CD19, CD20, CD23. Уровень поверхностных иммуноглобулинов CD20 и CD79b на опухолевых клетках ниже, чем на нормальных В-лимфоцитах.
Лимфому из малых лимфоцитов диагностируют при наличии лимфаденопатии, спленомегалии, цитопении при условии, что количество В-лимфоцитов в крови не превышает 5×10 9 /л. Обязательным условием для установления диагноза является биопсия лимфоузла [5].
По клиническому течению ХЛЛ крайне разнороден. Прогноз заболевания зависит от наличия или отсутствия неблагоприятных клинических, морфологических и молекулярно-генетических признаков. Гетерогенность клинико-лабораторных проявлений легла в основу классификации ХЛЛ, разработанной Воробьевым А.И. и Бриллиант М.Д. Выделяют восемь форм заболевания: 1) доброкачественную, 2) опухолевую, 3) прогрессирующую, 4) селезеночную, 5) костно-мозговую, 6) абдоминальную, 7) пролимфоцитарную и 8) лимфоплазмоцитарную [3].
К клинико-лабораторным прогностическим маркёрам относят лейкоцитоз периферической крови на момент начала терапии, время удвоения лимфоцитов, клиническую стадию заболевания, тип поражения костного мозга, пол, возраст, статус пациента по шкале ECOG и общий соматический статус [6]. Биологию опухоли отражают хромосомные аберрации, определяемые методом FISH, мутационный статус VH-генов, уровень экспрессии CD 38, ZAP 70. Наиболее распространёнными хромосомными изменениями являются del 13q14 (40-60 %), трисомия 12 хромосомы (15–30 %), del 17p13 (10%) [15].
Анализ кариотипа, клинических проявлений и длительности заболевания показал, что при изолированной делеции 13q наблюдается стабильное состояние и медленное прогрессирование с хорошим ответом на терапию. Прогностическое значение трисомии 12 хромосомы до настоящего момента является предметом дискуссий. Наличие 17p зачастую связано с мутацией гена-супрессора опухоли TP53 и ассоциировано с неблагоприятным течением болезни [35]. Медиана выживаемости больных с трисомией 12 составляет 114 месяцев, с делецией 11q - 79 месяцев, а с делецией 17p - 32 месяца [16].
Мутационный статус JgVH-генов при ХЛЛ отражает наиболее существенные биологические особенности заболевания. По результатам нескольких исследований обнаружено, что медиана выживаемости при современной терапии колеблется от 79 до 119 месяцев при отсутствии мутаций, в то время как при их наличии составляет 200–300 месяцев [29].
Первым маркёром, имеющим корреляцию с мутационным статусом, стал CD38+. Пороговый уровень экспрессии CD38+ на лимфоцитах в периферической крови при ХЛЛ составляет 30 % [5]. Медиана выживаемости в CD38-позитивной группе составила 109 месяцев, в СD38-негативной - 293 месяца [39]. Выявлена прямая взаимосвязь между отсутствием мутаций IgVH-генов и экспрессией тирозинкиназы ZAP-70, липопротеинлипазы (LPL) и металлопротеазы (ADAM29) [32]. Кроме вышеуказанных факторов, имеется не менее десятка других прогностических факторов, к которым можно отнести мутацию гена BCL6, экспрессию гена BCL2, уровень лактатдегидрогеназы (ЛДГ), β2-микроглобулин в сыворотке крови, экспрессию цитокинов (ИЛ-4, ИЛ-6) и фактора роста эндотелия сосудов (VEGF).
Выбор терапии и время начала лечения зависит от возраста, сопутствующей патологии, наличия факторов неблагоприятного прогноза.
В свою очередь, эффективность терапии во многом зависит от соблюдения дозы и режима введения химиопрепаратов. Цель терапии при ХЛЛ – достижение ремиссии. Клинико-гематологические критерии ответа на лечение при В-ХЛЛ не позволяют судить о глубине ремиссии, т. е. о количестве остающихся лимфоцитов В-ХЛЛ в крови и костном мозге больного. Было доказано, чем ниже уровень остающихся опухолевых клеток В-ХЛЛ, определяемый проточной цитометрией или полимеразной цепной реакцией (ПЦР), тем лучше прогноз заболевания (более длительная ВБП и ОВ). Доказано, что полуколичественные методы определения минимальной остаточной болезни (МОБ) не подходят для клиники. В настоящее время наиболее доступным и широко используемым методом для определения МОБ стала четырехцветная проточная цитометрия.
Первыми препаратами лечения хронического лимфоидного лейкоза являлись уретан, соли мышьяка. С 1905 года и последующие 50 лет основным методом лечения была локальная рентгенотерапия. В настоящий момент лучевая терапия практически не используется. В 1953 году появились алкилирующие препараты, первым из которых стал эмбихин, в настоящий момент используется хлорамбуцил [2]. Согласно результатам исследований не выявлено различий в выживаемости при раннем и отсроченном назначении хлорамбуцила [37]. Вслед за хлорамбуцилом были синтезированы новые алкилирующие препараты, до настоящего момента используют только циклофосфамид (С). Третьим важнейшим этапом в терапии ХЛЛ стало создание пуриновых аналогов. Одним из первых был синтезирован флударабин (F). Практически одновременно получены другие пуриновые аналоги - пентостатин, кладрибин [2].
Первые клинические испытания были проведены у ранее леченных больных и показали высокую эффективность использования флударабина. У 45 % рефрактерных ко всей терапии больных была получена ремиссия: у 13 % – полная ремиссия (ПР) продолжительностью 21 месяц, у 32 % - частичная ремиссия (ЧР) продолжительностью 13 месяцев [27]. Международной рабочей группой по ХЛЛ, исследовавшей 695 больных, оценивалась эффективность СНОР, САР и флударабина у пациентов в стадиях В и С. ПР и ЧР были достигнуты у 66 % больных, получавших САР, у 77 %, получавших СНОР, и у 81 % после лечения флударабином. Число ПР составило 13, 28 и 37 % соответственно. Медиана выживаемости составила 70 месяцев при лечении по схеме САР, 68 месяцев при терапии по программе СНОР, 74 месяца при лечении флударабином [14].
С конца 90-х годов в клинической практике активно используется моноклональное антитело к антигену CD20 – ритуксимаб (R). У ранее не леченных пациентов общий ответ (ОО) от терапии ритуксимабом в монорежиме составляет 51-86 %, частота ПР 4-19 %. В группе, получавшей лечение ранее, общий ответ получен у 25-45 % больных, ПР лишь у 3 %.
Результаты второй фазы рандомизированного исследования эффективности флударабина с ритуксимабом (RF) в различных режимах доказали, что сочетанное применение RF у ранее не леченных больных позволяет достичь полных и частичных клинико-гематологических ремиссий в 90 % случаев, из них в 47 % – ПР. При лечении флударабином в течение 6 месяцев, с последующим назначением ритуксимаба в течение 2 месяцев, получено лишь 77 % ремиссий, из них 28 % ПР. При медиане наблюдения 23 месяца в обеих группах не была достигнута медиана как безрецидивной, так и общей выживаемости [11].
Высокоэффективными оказались комбинации флударабина с митоксантроном (М) (77 % ремиссий, из них 20 % - ПР), эпирубицином (92 % ремиссий, 40 % - ПР) и циклофосфамидом (88–100 % ремиссий, 35–50 % - ПР).
Согласно исследованию испанской группы, включающих 69 пациентов в возрасте до 65 лет с впервые диагностированным ХЛЛ, к флударабину и циклофосфану добавляли митоксантрон. Общий ответ составил 90 %, МОБ-отрицательный полный ответ составил 26 %, МОБ-положительный полный ответ 38 %, ЧР - 26 %. Тяжелая нейтропения (3 или 4 степени) развилась у 10 % пациентов. Инфекционные осложнения были зарегистрированы в 9 % случаев, соответственно. Медиана продолжительности ответа составила 37 месяцев. У пациентов с делецией 17p ПР достичь не удалось [8].
В одном из исследований оценено сочетанное применение флударабина в сочетании с разными дозами циклофосфамида и митоксантрона у 60 пациентов с рецидивирующей или резистентной формой ХЛЛ. Полный ответ получен у 30 пациентов (50 %), из них 10 случаев (17 %) - с МОБ-отрицательным, и 17 (28 %) с частичным ответом. Средняя продолжительность ответа составила 19 месяцев. Основными осложнениями являлись инфекции – 8 %, нейтропения, тошнота и рвота. На фоне лечения летальность от инфекционных осложнений составила 5 % [9].
Некоторые исследователи пытались получить более высокий показатель ремиссии, добавляя дексаметазон (D) к флударабину и циклофосфамиду. Большое количество работ посвящено FMD-режиму, применение которого позволяет получить до 94 % ответов (47 % ПР), даже у пациентов, леченных ранее интенсивной терапией. А при использовании данного протокола в качестве терапии I линии частота достижения ПР оказалась еще выше (79 %) при общем ответе у 95 % больных. Чрезвычайно важно обратить внимание на то, что при достижении ПР в 82 % случаев регистрировались молекулярные ремиссии и в 84 % ПР сохранялись в течение 2-х лет. Из побочных эффектов данной комбинации отмечались лишь оппортунистические инфекции [1].
Группой исследователей из Барселоны использовалось сочетание химиопрепаратов флударабин, циклофосфамид, митоксантрон, ритуксимаб (R-FСМ). Было проведено шесть курсов терапии. Достигшие ответа пациенты в качестве поддерживающей терапии получали ритуксимаб каждые 4 месяца. ОО, ПР при отсутствии МОБ, ПР при наличии МОБ, ЧР - составили 93 %, 46 %, 36 % и 11 % соответственно. Тяжелая нейтропения развивалась у 13 % пациентов. Большие и малые инфекции были зарегистрированы у 8 % и 5 % пациентов соответственно. Установлено, что более поздние стадии, делеция 17p или высокий уровень бета-2-микроглобулина в сыворотке крови коррелируют с более низкой вероятностью достижения ПР [10].
В 2010 году опубликованы данные российских ученых, где была дана оценка эффективности различных режимов терапии ХЛЛ, включающих флударабин (RFC, FCM, FC). В исследование было включено 229 больных, из них 78 получали программу RFC, 72 – FCM, 79 – FC. В результате применения комбинации RFC клинически значимый лечебный эффект получен у 96 % больных, ПР у 80 % первичных больных и у 53 % ранее леченных пациентов. При назначении программы FCM положительный ответ отмечен у 93 % больных, ПР - у 75 % первичных и у 42 % ранее леченных пациентов. При лечении FC общий эффект составил 80 %, ПР наблюдалась у 41 % первичных и у 14 % ранее леченных больных. Сравнительный анализ ответа на терапию показал, что результативность комбинации RFC достоверно превышает эффективность программ FCM и FC без повышения токсичности, что позволяет рассматривать режим RFC в качестве программы выбора в терапии ХЛЛ [4].
Комбинированная терапия с флударабином, циклофосфамидом и ритуксимабом (FCR) в настоящее время является стандартом первой линии лечения ХЛЛ. Однако из-за токсичности режим FCR может применяться только у пациентов без выраженных сопутствующих заболеваний [17].
Не нужно забывать, что ХЛЛ является болезнью пожилых людей и зачастую сопутствующая патология и тяжёлые осложнения могут стать препятствием к назначению терапии RFC. Выбирая курс, необходим взвешенный подход, ведь в настоящий момент данное заболевание является не излечимым, нужно соблюдать баланс между токсичностью и эффективностью.
Одним из подходов к терапии пожилых больных, страдающих ХЛЛ, является применение курса RFC в редуцированных дозах (RFClite). В одном из исследований проводили терапию RFC lite 50 пациентам в возрасте 58 лет. Средняя продолжительность ответа составила 22,3 месяца (5,2–42,5). Нейтропения III–IV степени была отмечена в 13 % случаев во время проведения циклов ПХТ [20]. Кроме того, для пожилых больных можно использовать терапию лейкеран (хлорамбуцил) в комбинации с ритуксимабом. В исследование данной схемы было включено 100 пациентов, средний возраст 70 лет (43–86), медиана наблюдения 30 месяцев. Общий ответ составил 84 %, ПР достигнуты в 10 %. Терапия R-хлорамбуцил увеличивает частоту ответа в большей степени, чем хлорамбуцил в монорежиме, хотя ремиссии достигаются в меньшей степени, чем при RFC [24].
Большое количество работ посвящено назначению к химиотерапии интерферонов, что способствует увеличению безрецидивной выживаемости у пациентов с индолентными лимфомами, к которым можно отнести лимфому из малых лимфоцитов. Общий ответ у пациентов, которым ранее не проводили терапию по поводу основного заболевания, составил 75 %, у ранее леченных больных - 76 %. Медиана до прогрессии заболевания составила 12 месяцев. Токсичность III степени проявлялась нейтропенией у 39 % больных, анемией - у 17 %, тромбоцитопений - у 5 % [30].
С 2008 года в США одобрен к применению лекарственный препарат бендамустин, обладающий бифункциональной алкилирующей активностью и антиметаболическими свойствами пуриновых аналогов. На фоне лечения бендамустином отмечено улучшение показателей частоты объективного ответа (68 % против 31%) и полного ответа (31 % против 2 %) по сравнению с хлорамбуцилом. Также на фоне терапии бендамустином отмечено значительное увеличение ВБП по сравнению с хлорамбуцилом (21,6 против 8,3 месяцев) [28].
Доказана высокая эффективность бендамустина в комбинации с Ритуксимабом (протокол CLL2М) [19]. Общий ответ на терапию составил 90,9 %: у 36 (32,7 %) больных наблюдалась ПР, у 61 (55,5 %) - ЧР и у 3 (2,7 %) - нодальная ЧР. У 10 (9,1 %) пациентов была зарегистрирована стабилизация заболевания. Однако среди 7 пациентов с делецией 17p только у 3 (42,9 %) была получена ЧР.
Продолжением этой работы служит многоцентровое открытое рандомизированное исследование III фазы CLL10, в котором комбинация BR сравнивается со стандартом терапии первой линии FCR. Всего в исследование включено 688 пациента без делеции 17p. Данная работа подтвердила преимущество эффективности терапии FCR, где были более высокие показатели ПР, беспрогрессивной и бессобытийной выживаемости. Преимуществом программы BR стало меньшее количество инфекционных осложнений, что особенно важно для ослабленных и пожилых больных [17].
Серьёзной проблемой в терапии ХЛЛ остаётся рефрактерность к флударабину, которая ассоциируется с резистентностью к другим цитостатикам и низкой медианой ОВ, не превышающей 1–2 лет.
Алемтузумаб (Кэмпас), представляющий собой анти-CD52 антитела, применялся в лечении резистентных форм ХЛЛ. Алемтузумаб индуцирует клинический ответ у 40 % пациентов с рефрактерным течением ХЛЛ и у 80 % пациентов при использовании в терапии первой линии [33]. В отличие от большинства других схем при ХЛЛ алемтузумаб оказался одинаково эффективен у пациентов с делецией 17р хромосомы по сравнению с другими цитогенетическими подгруппами. Добавление алемтузумаба к программе FCR способствовало достижению быстрого ответа у пациентов с рецидивирующим течением ХЛЛ, но сопровождалось большим количеством инфекционных осложнений как во время проведения терапии, так и после ее окончания [7].
В течение пяти последних лет появились моноклональные анти-СD20 антитела нового поколения. Хорошие результаты в лечении ХЛЛ получены при использовании офатумумаба. Офатомумаб – это полностью человеческое моноклональное антитело к CD20 антигену, обладающее способностью ингибировать раннюю активацию B-лимфоцитов. В международном исследовании 138 больных были разделены на две группы: 59 человек, резистентных к флударабину и алемтузумабу (FA-реф.), 79 - устойчивых к флударабину (BF-реф.), и наличием противопоказаний к назначению алемтузумаба. Группа BF-реф. характеризовалась большой опухолевой массой (лимфатические узлы более 5 см). Общий процент ответа составил 58 % в первой группе и 47 % второй. Полная регрессия симптомов заболевания и улучшение общего самочувствия достигнуты в 57 % и 48 % случаев соответственно. Медиана выживаемости без прогрессирования и ОВ составила 5,7 и 13,7 месяцев в FA-реф. группе, и 5,9 и 15,4 месяцев в BF-реф. группе, соответственно [41]. Обинутузумаб – глико-инженерное гуманизированное моноклональное антитело II типа, которое специфически связывается с особым белком-антигеном CD20 на поверхности злокачественных В-лимфоцитов.
Читайте также: