
Хромосомные транслокации при лейкозе
Транслокация (4;11)(q21;q23) — специфическая хромосомная транслокация, которую удается обнаружить при различных вариантах острого лейкоза. Чаще всего она встречается при остром лимфобластном лейкозе и бифенотипических острых лейкозах, но наблюдается и при миелоидных острых лейкозах, преимущественно монобластных и миеломонобластных.
Эта транслокация наиболее характерна для детей в возрасте до 1 года (более 50 % от общего количества острых лейкозов). При лейкозах у детей до 6 мес она обнаруживается еще чаще, а у детей старше 1 года и у взрослых с ОЛЛ ее частота составляет 10—15 %. Клинически лейкозы с t(4;11) характеризуются очень высоким лейкоцитозом и большим числом бластов, а также выраженной опухолевой инфильтрацией органов (гепатоспленомегалия, лимфаденопатия).
У подавляющего большинства больных лейкемические бласты можно отнести к про-В- или пре-В-клеткам. Нередко наблюдается коэкспрессия миелоидных антигенов, в частности CD65s.
При этой транслокации происходит слияние фрагментов двух генов — MLL (11q23) и AF4 (4q21), в результате чего образуются химерные гены MLL-AF4 и AF4-MLL. Принято считать, что химерный ген MLL-AF4, сформировавшийся на хромосоме 11, играет решающую роль в развитии лейкоза. Разработаны очень чувствительные молекулярные зонды, позволяющие выявлять практически все перестройки гена MLL с помощью методов PCR и FISH.
Лейкозы с t(4; 11) или с другими транслокациями, в которых участвует хромосомный район 11q23 (ген MLL), в большинстве своем неблагоприятны в прогностическом отношении. У взрослых пациентов прогноз практически такой же, как при Ph-транслокации, у детей до 1 года еще хуже. Наихудший прогноз отмечен у детей до 6 мес. Так, медиана безрецидивного течения ОЛЛ с t(4;11) у детей до 1 года составляет 7 мес, от 1 года до 10 лет прогноз значительно лучше (возможна многолетняя полная ремиссия), а у детей старше 10 лет он опять ухудшается.
Иными словами, эффективность лечения больных острым лимфобластным лейкозом с t(4;11) у детей от 1 года до 9 лет значительно выше, чем у взрослых.
![]()
Известны транслокации района 11q23 более чем с 70 различными участками хромосом. Они встречаются с разной частотой. Например, t(9;11) и t(11;19) наблюдаются чаще, чем t(6:11), t(10;11).
Необходимо учитывать следующие важные моменты:
1. Обнаружение перестроек хромосомного района llq23 не всегда легко; в ряде случаев это не удается сделать при стандартном хромосомном анализе и требует применения молекулярно-генетических методов. В частности, при таких транслокациях, как t(9;11), t(6:11) и t(11;19), происходит обмен небольшими фрагментами хромосом, и стандартный цитогенетический анализ позволяет их обнаружить только на препаратах высокого качества. Более информативны FISH и ПЦР.
Был изучен костный мозг 170 больных острым лимфобластным лейкозом (100 взрослых и 70 детей) с использованием как хромосомного анализа, так и ПЦР (так называемая multiplex PCR). У 41 пациента кариотип был нормальным, но у 13 из них, т. е. практически у каждого третьего (!), выявлены аномалии при проверке с некоторыми специфичными (для острого лимфобластного лейкоза транслокаций) праймерами. У 4 из 13 больных обнаружены перестройки гена MLL.
2. Цитогенетические изменения, выглядящие при световой микроскопии абсолютно идентичными, оказываются различными при использовании молекулярно-генетических методик. Установлено, в частности, что t(10;11)(p13;q23) и t(11;19)(q23;p13) могут затрагивать различные гены и обусловливать различия в чувствительности к химиопрепаратам.
Изменения гена MLL, как правило, имеют неблагоприятное прогностическое значение, поэтому их выявление с помощью FISH и ПЦР настоятельно рекомендуется при остром лимфобластном лейкозе с нормальным кариотипом или при сложных структурных перестройках хромосом. В этих ситуациях можно обнаружить субмикроскопическую дупликацию или амплификацию гена MLL.
Стандартный хромосомный анализ может оказаться неинформативным вследствие низкого митотического потенциала лейкозных клеток. В таких случаях цитогенетик видит только делящиеся нелейкозные (нормальные) клетки. В гематологических клиниках, где методы FISH и ПЦР вошли в рутинный диагностический набор, изменения гена MLL выявляются значительно чаще, чем в слабооснащенных клиниках.
Транслокация t (1; 19) (q23; p13), которая приводит к химерному генам TCF3-PBX1, является одной из наиболее частых перегруппировок, наблюдаемых при острой лимфобластной лейкемии В-клеток. Он проявляется как у взрослых, так и у детей с острой лимфобластной лейкемией В-клеток на общей частоте от 3 до 5%. В большинстве случаев острый лимфобластный лейкоз пред-В-клеток, несущий транслокацию t (1; 19), имеет типичный иммунофенотип с гомогенной экспрессией CD19, CD10, CD9, полным отсутствием CD34 и по меньшей мере уменьшенным CD20. Кроме того, транслокация t (1; 19) коррелирует с известными клиническими факторами высокого риска, такими как повышенное количество лейкоцитов, высокий уровень лактатдегидрогеназы в сыворотке и вовлечение центральной нервной системы; ранние сообщения показали, что пациенты с транслокацией t (1; 19) имели низкий исход при стандартном лечении.
Мы сообщим о случае 15-летнего сирийского мальчика с острой лимфобластной лейкемией пред-В-клеток с аномальным кариотипом с der (19) t (1; 19) (q21,1, p13,3) и двух еще не зарегистрированных хромосомных аберрации: интерстициальная делеция 6q12 — 6q26 и der der (13) t (1; 13) (q21.1; p13). Согласно литературе, случаи, у которых транслокация t (1; 19) -положительная, имеют значительно более высокий уровень рецидива центральной нервной системы, чем у пациентов с острой лимфобластной лейкемией без транслокации. Интересно, что участие нашей центральной нервной системы также наблюдалось у нашего пациента.
Насколько нам известно, это первый случай острого лимфобластного лейкоза детского пред-В-клеток с несбалансированной транслокацией t (1; 19) с двумя дополнительными хромосомными аберрациями, del (6) (q12q26) и t (1; 13 ) (q21.3; p13), которые, как представляется, повторяются и могут влиять на клинический результат. Также в настоящем случае подтверждается влияние транслокации t (1; 19) на рецидив центральной нервной системы, который необходимо изучить для будущих механизмов в будущем.
Транслокация t (1; 19) (q23; p13) представляет собой неоднократно, но в целом редко наблюдаемую перегруппировку в остром лимфобластном лейкемии В-клеток (B-ALL) и может быть обнаружена у взрослых и педиатрических пациентов с общей частотой от 3 до 5 % [1, 2]. Большинство случаев pre-B-ALL, несущих транслокацию t (1; 19), экспрессируют типичный иммунофенотип с гомогенной экспрессией CD19, CD10, CD9, полным отсутствием CD34 и сниженной экспрессией CD20 [3]. Эта транслокация может происходить в двух формах: сбалансированной или несбалансированной. Несбалансированная форма pre-B-ALL приводит к производной хромосомы 19 [2]. Как сбалансированная, так и несбалансированная транслокации t (1; 19) приводят к слиянию транскрипционного фактора 3 (TCF3), расположенного в 19p13, с дообедонным лейкозом 1 в раннем возрасте (PBX1) в 1q23, с образованием химерного гена, белковый продукт которого изменяется , среди других клеточных процессов, остановка дифференцировки клеток [1]. Конкретно, ген слияния кодирует транскрипционный фактор, несущий трансактивационный домен TCF3 и домен связывания дезоксирибонуклеиновой кислоты (ДНК) PBX1, что облегчает активацию генов конституции [4]. Кроме того, слитый белок, по-видимому, оказывает доминирующее отрицательное влияние на активность TCF3 дикого типа. Таким образом, как увеличение экспрессии генов-мишеней PBX1, так и снижение активности TCF3 считаются важными в лейкемогенезе [5].
Другие гены партнера TCF3 включают ZNF384 (12p13, неизвестный прогноз), NOL1 (12p13, неизвестный прогноз), неизвестный ген партнера в 13q14 (прогноз неизвестен), HLF (17q22, крайне плохой прогноз) и FB1 / TFPT (19q13.4 прогноз неизвестен) 6. Кроме того, транслокация t (1; 19) коррелирует с известными клиническими особенностями высокого риска, такими как повышенное количество лейкоцитов (WBC), высокий уровень лактатдегидрогеназы в сыворотке (LDH), вовлечение центральной нервной системы (ЦНС) и плохой результат в стандартное лечение [9].
Здесь мы сообщаем результаты клинических, G-полосчатых и молекулярно-цитогенетических результатов, полученных у детского пациента с пред-B-ALL с несбалансированной транслокацией t (1; 19) и двумя дополнительными хромосомными аберрациями.
Он был передан (25 мая 2014 г.) в нашу лабораторию лабораторий хромосом и лабораторию проточной цитометрии, кафедру молекулярной биологии и биотехнологии Комиссии по атомной энергии Сирии для цитогенетики и проточно-цитометрического анализа. Его можно было бы отнести к группе высокого риска. Таким образом, после первоначального диагноза в течение 5 месяцев он был снабжен немецкой исследовательской группой Multicenter Study для лечения острого лимфобластного лейкоза (GMALL). К сожалению, из-за политической ситуации в его родной стране ему были предоставлены доступные лекарства, такие как винкристин 1,4 мг / м2, доксорубицин 25 мг / м2 и метотрексат 20 мг / м2. Он ответил на это лечение без каких-либо проникновений в свой БМ; он много раз получал тромбоциты и переливание крови; его ПБ показал панцитопению и нейтропению. Лечение останавливали до фазы консолидации, потому что его оценка биопсии BM показала морфологическую ремиссию (8% бластов осталось); Тест спинномозговой жидкости показал аномальные клетки; у него также была легочная инфекция. Примерно через 5,5 месяца после первоначального диагноза он умер по неизвестным причинам во время лечения. Никакого вскрытия не было, потому что он умер в своем доме. Его родители согласились с научной оценкой дела, и исследование было одобрено этическим комитетом Комиссии по атомной энергии, Дамаск, Сирия.
Анализ хромосом на образце BM с использованием GTG-обвязки в соответствии со стандартными процедурами [10] был выполнен до начала лечения; он показал кариотип 46, XY, del (6) (q?), der (13) t (1; 13), der (19) t (1; 19) [6] / 46, XY, del (6 ) (д), дер (19) т (1;? 19) [5] / 47, XY, + I (1) (д), дель (6) (д), дер (19) т (1? , 19) [1] / 46, XY [6] (рис.1). Кариотип был описан в соответствии с Международной системой цитогенетической номенклатуры человека (ISCN) 2013 года [11]. 1GTG-banding выявил следующий кариотип в 6/18 метафазах: 46, XY, del (6) (q?), Der (13) t (1; 13), der (19) t (1; 19) [6] , Все производные хромосомы отмечены и выделены стрелочными головками
Дальнейшие исследования проводились на образце BM с использованием молекулярной цитогенетики (фиг.2). Мы провели двухцветную флуоресцентную гибридизацию in situ (D-FISH) с помощью зондов цельной хромосомной краски (WCP) для хромосом 1, 6, 13 и 19 (MetaSystems, Altlussheim, Germany) [10], которые не предоставили никакой информации на криптографических транслокациях (данные не показаны) и проверенной массивом многоцветной полосы с высоким разрешением (aMCB) [12], с использованием зондов для соответствующих хромосом 1, 6, 13 и 19, участвующих в соответствии с GTG-обвязкой (фиг.2 ). Обратную транскриптазо-полимеразную цепную реакцию (RT-PCR) для транскриптов слияния E2A-PBX1 проводили перед обработкой с использованием специфических праймеров, ранее описанных [13], подтвердили наличие слияния TCF3 / PBX1 (транскрипт E2A / PBX1, 373-base полоса), чаще всего выявляемая при остром лимфобластном лейкозе (ВСЕ, данные не показаны). Таким образом, следующий конечный кариотип перед лечением определяли с использованием флуоресцентного микроскопа (AxioImager.Z1 mot, Carl Zeiss Ltd, Hertfordshire, UK), оборудованного соответствующими наборами фильтров для различения максимум пяти флуорохромов плюс встречное пятно 4 ‘, 6- диамидино-2-фенилиндола (DAPI). Захват и обработка изображений выполнялись с использованием системы визуализации ISIS (MetaSystems): 46, XY, del (6) (q12q26), der (13) t (1; 13) (q21.1; p13), der (19) t (1; 19) (q21.1; p13.3) [6] / 46, XY, дель (6) (q12q26), дер (19) т (1; 19) (q21.1; p13.3) [ 5] / 47, XY, + I (1) (q10), дель (6) (q12q26), дер (19) т (1; 19) (q21.1; p13.3) [1] / 46, XY [6] .Fig. Показаны результаты двухцветных диапазонов с подтверждением массива. Нормальные хромосомы (#) изображены с левой стороны каждого изображения и производная четырех хромосом на правой стороне нормальных хромосом. Неокрашенные области при подаче на хромосомы, специфичные для массива, многоцветные зондирующие зонды на производных хромосомах, показаны серым цветом. Различные описания псевдоколора для массива с проверенным множеством многоцветных зондов1 выявили точки останова в производных хромосомах 13, 19 и изохромосоме 1q. b Проверенная временем многоцветная полоса 6 обнаружила интерстициальное удаление в der (6). c Проверенная множителем многоцветная полоса 13 показала, что практически вся короткая рука хромосомы 13 осталась нетронутой в дер (13). d Точка останова на der (19) может быть определена с помощью многозонного набора зондов19. # хромосома, производная хромосома, многоцветная полоса MCB
Иммунофенотипирование проводили на образце BM с использованием общей панели флуоресцентных антител против следующих антигенов, типичных для разных клеточных линий и типов клеток: CD1a, CD2, CD3, CD4, CD5, CD8, CD10, CD11b, CD11c, CD13, CD14, CD15, CD16, CD19, CD20, CD22, CD23, CD32, CD33, CD34, CD38, CD41a, CD45, CD56, CD57, CD64, CD103, CD117, CD123, CD138, CD209, CD235a и CD243; кроме того, были испытаны антитела к каппа и лямбда-легкие цепи, IgD, поверхностно-связанный IgM (sIgM) и связанный с человеческим лейкоцитарным антиген-антиген D (HLA-Dr). Все антитела были приобретены у BD Biosciences (San Jose CA, USA). Образцы анализировали на проточном цитометре BD FACSCalibur ™. Были включены автофлуоресценция, жизнеспособность и контроль изотипа. С помощью программного обеспечения BD Cellquest ™ Pro было проведено исследование и анализ потока цитометрических данных. Проточный цитометрический анализ образца BM характеризовал этот случай как пред-B-ALL в соответствии с классификациями ВОЗ. Аномальная популяция клеток (94% тестируемых клеток) была положительной для CD45 + dim, CD19 +, CD10 +, HLA-DR + и CD79a +. Эта клеточная популяция была отрицательной для CD34, CD2, CD7, CD13, CD14, CD15, CD20, CD33 и CD117.
Согласно литературе, транслокация t (1; 19) (q23; p13) представляет собой транслокацию, которая неоднократно, но в целом редко встречается в случаях B-ALL; он генерирует ген слияния TCF3-PBX1 [1, 2]. Согласно базе данных Мительмана об хромосомных аберрациях в раке [14], существует пять случаев ALL с транслокацией t (1; 19) (q21; p13), семь таких случаев с транслокацией t (1; 13) с короткими и / или длинными плечи обеих хромосом, пять случаев ALL с изохромосомой i (1) (q10) и 710 случаев ALLs с del (6) (q), включая 21 случай с del (6) (q12) и три случая с del (6 ) (Q26). Кроме того, хромосомные полосы 1q21, 13p13 и 19p13 участвуют в хромосомных перестройках в 252, 785 и 8 случаях соответственно [14]. Насколько нам известно, настоящий отчет о заболевании является первым, кто наблюдает за детским пред-B-ALL с несбалансированной транслокацией t (1; 19) (q21.1, p13.3), связанной с двумя дополнительными хромосомными аберрациями del ( 6) (q12q26) и der (13) t (1; 13) (q21.1; p13) [14]. Интересно, что одна метафаза с изохромосомой 1q также наблюдалась в GTG-обвязке и обнаруживалась в 1/25 метафазах при анализе флуоресценции in situ-гибридизации (FISH) (рис.2). Как упоминалось выше, это было также замечено в пяти предыдущих случаях ALL [14].
Транслокация t (1; 19) (q23; p13) наблюдается в двух основных формах [2, 15, 16]. Известно, что аномалии хромосомы 1, такие как полная или частичная трисомия для длинного плеча, возникают во время эволюции клона и могут наблюдаться как повторяющиеся при гематологических злокачественных новообразованиях 18.
Ген CKS1B, нанесенный на карту 1q21, является членом семейства высококонсервативных циклинкиназных субъединиц 1 (CKS1), которое взаимодействует с циклинзависимыми киназами (Cdks) и играет критическую роль в прогрессировании клеточного цикла [19]. Сверхэкспрессия CKS1B коррелирует с низкой экспрессией p27 и неблагоприятной выживаемостью в нескольких злокачественных новообразованиях человека, включая рак желудка, колоректального и устного плоскоклеточного рака 21 из-за его критической роли регулятора клеточного цикла и его участия в различных карциномах человека.
Эта область 6q12-22 содержит несколько генов с известной или предполагаемой функцией подавления опухолей, включая MAP3K7, для которой недавно была продемонстрирована роль подавляющей опухоль для эпителиальных клеток предстательной железы [23]. MAP3K7 является вездесущим, выраженным и участвующим в различных биологических процессах, таких как рост клеток, дифференцировка и апоптоз. MAP3K7 оказывает эти эффекты, взаимодействуя с несколькими различными сигнальными путями и молекулами-активаторами 24.
В отличие от ранних сообщений о пациентах с транслокацией t (1; 19), сообщающих о более неблагоприятном исходе для пациентов со сбалансированной транслокацией по сравнению с пациентами с несбалансированной транслокацией t (1; 19) 28, более поздние исследования не обнаружили разницы в выживаемости без событий (EFS) среди этих двух подгрупп 32. Однако случаи, у которых транслокация t (1; 19) -положительная, имеют значительно более высокий уровень участия ЦНС в случаях рецидива, чем у пациентов со ВСЕМИ без транслокации [30]; группа Св. Иуды наблюдала четырех пациентов с рецидивом ЦНС, но не у пациентов с рецидивом БМ среди 41 пациента с транслокацией t (1; 19) [33]. Кроме того, Moorman et al. [34] сообщили о шести рецидивах у 50 пациентов с транслокацией t (1; 19), из которых три рецидива включали ЦНС. Более того, было установлено, что заболевание ЦНС чаще встречается у пациентов с Т-клеточной острой лимфобластной лейкемией (T-ALL) по сравнению с пациентами с пред-B-ALL [35], а участие ЦНС связано с плохой выживаемостью. Fielding et al. [36] также показали, что исход рецидивирующих пациентов был очень слабым, и что у пациентов с Т-клеточным заболеванием наблюдалась 5-летняя выживаемость на 5% по сравнению с 8% у пациентов с В-ALL.
Дополнительные цитогенетические аномалии (ACAs) наблюдались у 63% (47 из 2640, что составляет 1,8%, у детей с диагнозом B-ALL) без существенных различий между транслокацией t (1, 19) и / или der (19) t (1; 19) -положительные случаи [37]. Наиболее распространенными ACAs были del (9p), i (9q), del (6q) и del (13q) [16, 20]. Ни один из их 47 пациентов с транслокацией t (1; 19) не продемонстрировал сопутствующих аберраций 13p13 и / или 6q12 до 6q26 областей. Таким образом, клиническая значимость несбалансированной транслокации t (1; 19) (q21.1, p13.3) у нашего пациента с транслокацией 13p13 и / или del (6) (q12q26) неясна. Согласно литературе, присутствие ACA не оказывает существенного влияния на EFS или общую выживаемость [35]. Это наблюдение согласуется с выводами других рецидивирующих связанных с лейкемией транслокаций, где ACAs, по-видимому, не влияют на прогноз [37, 38]. Тем не менее, неблагоприятный исход настоящего случая может быть намеком на то, что есть исключения из этого правила.
Пациенты с педиатрическим ALL с числом WBC более 50 × 109 / л считаются подверженными высокому риску рецидива и, таким образом, получают интенсивное лечение [39, 40]. При ретроспективном анализе пациенты с гиперлейкоцитозом (количество WBC> 50 × 109 / л) были достоверно коррелированы с более коротким временем выживания. Цитогенетические особенности были тесно связаны с WBC и по крайней мере частично объясняли прогностическую ценность WBC, хотя есть данные о том, что у детей с подобными цитогенетическими аберрациями могут быть очень разные WBC 41.
Здесь мы описали наличие двух необычных хромосомных аберраций, т. Е. Del (6) (q12q26) и der (13) t (1; 13) (q21.1; p13) в случае детского пред-B-ALL с несбалансированной транслокацией t (1; 19), что может указывать на то, что специальные ACA могут отрицательно влиять на клинические исходы, в отличие от того, что широко обсуждается в настоящее время.
Дополнительные цитогенетические аномалии
Острый лимфобластный лейкоз
Многоцветная полоса с высокой разрешающей способностью

Мы продолжаем обсуждение темы лейкозов у детей, и поговорим еще о некоторых нюансах в их диагностике и лечении. Были уже обсуждены вопросы их возникновения и диагностики, но пока мы еще не дошли до вопросов обсуждения лечебной тактики и реабилитации. Продолжим обсуждение.
Возможно ли предотвратить лейкозы?
Ранее лейкозы считались абсолютно летальными заболеваниями, от них не было лекарств и спасения, они практически во всех случаях приводили к гибели детей. На сегодня подходы к лечению и профилактике лейкозов изменились и многие из видов онкологических патологий можно предотвратить, если активно изменить свой образ жизни и образ жизни ребенка, что будет приводить к снижению влияния некоторых из особенно значимых факторов риска. Но это только способы снизить влияние негативных факторов, но не стопроцентно предотвратить развитие лейкоза. В настоящее время не существует современных способов для полноценной профилактики большинства из известных науке опухолей в детском возрасте. У подавляющего большинства детей или взрослых с лейкозами нет точно известных для именно лейкоза факторов риска и поэтому не существует и разработанных рекомендаций для профилактики лейкозов.
У детей с заведомо известными факторами риска по развитию лейкозов, как например, при наличии у ребенка синдрома Ли-Фраумени или при наличии синдрома Дауна, проводится тщательный и регулярный контроль с периодическими наблюдениями. Частота возникновения лейкозов у малышей с подобными генетическими синдромами хотя и повышается в сравнении с общими данными по популяции, но однако, все-таки не сильно повышается в современных условиях наблюдения и диагностики. В результате применения химиотерапии или же лучевой терапии у детей или взрослых с опухолевыми процессами, а также в результате применения иммуносупрессивной терапии при проведении трансплантации органов с целью профилактирования из отторжения, также вероятен повышенный риск развития лейкозов.
Возможно ли выявление лейкозов в ранней стадии
В сегодняшних условиях развития медицины не существует особых и специальных методов для того, чтобы провести раннее выявление лейкозов. Самой правильной и лучшей стратегией для ранней диагностики лейкозов становится повышенное внимание к проявляющимся признакам и выявленным симптомам данного заболевания (о симптомах лейкоза мы уже говорили – они неспецифичны и похожи на многие болезни). Будет требоваться очень пристальное и тщательное наблюдение за такими детьми с известными из генетических отклонений, которые заведомо могут повышать риски по возникновению лейкозов. Также особенно тщательное наблюдение требуется за теми детьми, кто получал химиотерапию, подвергался лучевым воздействиям по поводу опухолей, а также за теми, кто был подвержен операции по трансплантации органов и затем получал активную иммуносупрессивную терапию препаратами.
Установление стадии лейкозов у детей
При подавляющем большинстве опухолевых процессов необходимо определение стадии процесса – это необходимо для того, чтобы точно идентифицировать распространенность процесса. В подавляющем большинстве случаев стадии опухоли зависят от ее размеров и степени распространенности в организме. Однако, у лейкозов выставляются стадии не таким образом, как в большинстве других опухолей. При лейкозе костный мозг поражается изначально наряду с периферической кровью. И основной проблемой при лейкозе является проникновение и распространение лейкозных клеток по печени и селезенке, поражение половых органов и лимфатических узлов, а также органов центральной нервной системы. Если сами лейкозные клетки проникают в область ЦНС в больших количествах, тогда их наличие можно выявить при проведении исследования спинномозговой жидкости (исследование ее под микроскопом). Для таких целей проводится спинномозговая пункция, ее проводят в рамках планового первичного обследования при подозрении на онкологию.
При обнаружении наличия лейкозных леток важно провести более интенсивное обследование и активное лечение. Также постоянно существуют и различия у разных больных с положительным и отрицательным ответом на проводимую терапию, и они тогда называются прогностическими факторами. Такие факторы важны для определения общего прогноза в развитии и течении лейкоза у детей, и они важны для выбора и степени интенсивности проводимой терапии. Важно также знать, что прогностические факторы особенно важны для больных с наличием острого лимфобластного лейкоза, чем для тех, кому диагностирован острый миелобластный лейкоз.
Выявление прогностических факторов у детей с наличием строго лимфобластного лейкоза
Всех детей с острым лимфобластным лейкозом можно подразделить на несколько отдельных категорий – низкий риск, стандартный риск, высокий риск и очень высокий риск. Чем выше группа риска лейкоза, тем активнее и интенсивнее должна быть терапия лейкозов. В общем, у детей с группой низкого риска прогнозы и исходы лейкоза гораздо оптимистичнее, чем у таких же больных с лейкозами очень высокого риска. Также важен возраст самого ребенка и изначальное, исходное количество лейкоцитов периферической крови – это одни из важнейших факторов для будущего прогноза лечения и выздоровления. Важно помнить о том, что многие детишки с наличием как одного, так и более неблагоприятных факторов прогноза могут быть вполне активно излечимы в современных клиниках.
Возраст к моменту диагностики также важен для прогнозов – у детишек возрастом от одного года до девяти лет прогнозы болезни обычно более оптимистичные, а вот у больных до года или же старше десяти лет будут относиться к категориям высокого риска болезни. Не менее важно и количество лейкоцитов у ребенка, так как у больных с острым лимфобластным лейкозом с наличием очень высокого количества лейкоцитов к моменту первичного диагностирования болезни (более 50 тыс лейкоцитов в мм3), будут относиться к категориям высокого риска, и будут нуждаться в проведении более агрессивной и активной терапии. Также считается, что влияет и пол ребенка – у девочек с наличием острого лимфобласного лейкоза вероятности на излечение несколько больше, чем у мальчиков.
Также влияют и расовые особенности детей на течение и прогноз лейкоза. Например, у детей афроамериканского происхождения и детей из латинской Америки при развитии лейкоза выздоровление происходит реже, в сравнении с другими расами. Также, увеличение селезенки или печени обычно связывают с высокими количествами лейкоцитов, что также существенно влияет на прогнозы. Немаловажным для прогноза при лейкозе будет и проведение иммунотипирования клеток – определения их принадлежности к той или иной группе. У детей с наличием в крови большого количества незрелых или полузрелых В-лимфоцитов прогнозы в лечении будут лучше в сравнении с наличием лейкоза с Т-клеточными или же зрелыми В-клеточными лейкозами. Важными при типировании будет определение количества хромосом, так как у больных будет высокая вероятность излечения лейкозов в том случае, если количество хромосом внутри лейкозных клеток повышено. Особенно хорошим будет прогноз при наличии дополнительных хромосом в четвертой или десятой паре.
У детей с наличием лейкоза с редуцированным (сниженным) количеством хромосом в сравнении с нормальным количеством хромосом, прогнозы по самому заболеванию и по излечению. Отдельным вариантом будет наличие хромосомных транслокаций – это перенос кусочков хромосомы с одной пары на другую. У детей с лейкозами, чьи лейкозные клетки обладают транслокациями, особенно между 21 и 12 парами хромосом, вероятности по излечению лейкоза выше, чем у аналогичных больных с транслокациями по девятой и 22 парами, а также между первой и 19-ой, или же четвертой и 11-ой. И кроме того, самыми важными для прогноза при лейкозе будут также ответы на лечение. Больные, которые достигли ремиссии – когда полностью отсутствуют признаки заболевания на протяжении 14 суток химиотерапии, будут иметь самые лучшие прогнозы в сравнении с детьми, имеющими другие формы острого лимфобластного лейкоза, которым будет нужно в дальнейшем проводить активное и более интенсивное лечение детей.
Важно определить все признаки лейкозов, которые будут сильно влиять на дальнейшие прогнозы в выживаемости и лечении, а также на стойкость ремиссии. Доктора сразу же стараются определить все факторы, чтобы сразу же начать терапию именно по той схеме, которая будет наиболее актуальной и подходящей, так как все препараты обладают высокой токсичностью и достаточно тяжелы, лишние дозы облучения и химии при относительно благоприятной форме лейкоза просто не нужны.
Филадельфийская хромосома характерна для ХМЛ или хронического миелоидного лейкоза, который является клональным новообразованием, развивающимся из кроветворных стволовых клеток.
Данная опухоль была первой, при которой обнаружили признаки характерного хромосомного маркера. Открытие в 1960 году в Филадельфии сделали американские исследователи D. A. Hungerford и P. C. Nowell, в связи в чем этот маркер назвали термином филадельфийская хромосома-Ph. И именно эта находка послужила началом клинической цитогенетики в онкологии.
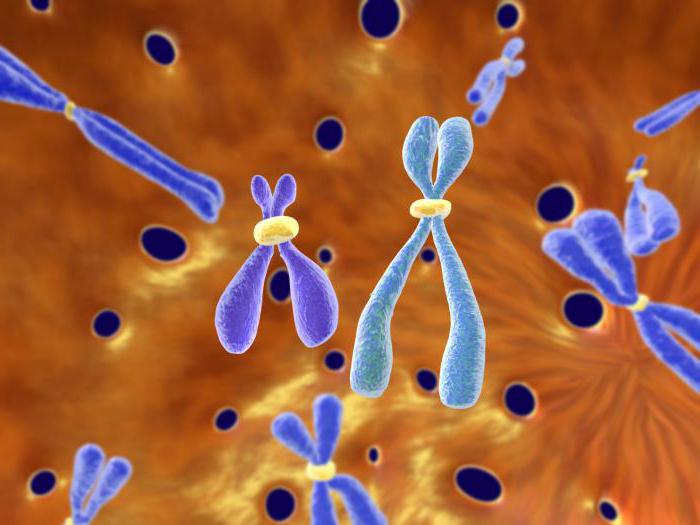
Что собой представляет хромосома?
Может появляться и при других видах лейкоза. Филадельфийская хромосома является укороченной хромосомой, входящей в группу малых акроцентриков. Каждая нормальная женская клетка этой группы содержит их 2 пары– 21 и 22, клетка же мужская включает не четыре, а пять таких хромосом, потому что помимо 21 и 22 пар включается еще и Y-хромосома.
Без G-бендинга, то есть при обычном окрасе, Ph-хромосома выявляется почти у каждого пациента, страдающего ХМЛ, а именно в 95-98 процентах случаев. На хромосомах, которые окрашены дифференциально, видно, что одна из 22 пар является укороченной.
Каков процент обнаружения?
Приблизительно у 90 % пациентов ее видно в каждой анализируемой метафазе, а у остальных больных обнаруживаются и клетки с хромосомой-Ph, и клетки нормальные.
К тому же в некоторых случаях Ph-хромосому регистрируют в меньшинстве клеток костного мозга. Транслокацию (9;22) наблюдают в мегакариоцитах, миелоидных клетках, В- и Т-лимфоцитах, эритробластах. Данный факт является свидетельством того, что болезнь берет начало с предшественницы гемопоэза – некоммитарованной клетки.
Атипичные транслокации
Приблизительно в 10 % случаев можно наблюдать атипичные транслокации, когда цитологическое стандартное исследование может позволить увидеть перенос части 22 хромосомы не на 9, а на любую другую. Более того, в некоторых случаях при ХМЛ обнаруживаются сложные Ph-транслокации при участии не 2 (22 и 9 хромосом), а 3 хромосом или большего количества.

Установили, что почти при каждой Ph-транслокации участвуют 9 и 22 хромосомы, но не всегда это можно увидеть при проведении стандартного цитогенетического исследования, однако это обнаруживают во время использования ПЦР и FISH.
Многочисленные исследователи полагают, что тип Ph-транслокации (сложная, атипичная, стандартная) клинического значения не имеет.
Разрыв генов
Использование методов молекулярно-генетических поспособствовало установлению того, что разрыв в 9-й хромосоме проходит через протоонкоген (ген ABL), который раньше был идентифицирован у мышей в одном из вирусов лейкоза. В 22-й хромосоме наблюдают разрыв BCR гена. Результатом слияния фрагментов генов BCR и ABL становится образование химерного гена BCR-ABL расположенного обычно на 22-й делетированной хромосоме.
Как вызывает филадельфийская хромосома лейкоз?
Приблизительно 70 % случаев характеризуется обнаружением помимо транскрипта BCR-ABL продукта другого химерного гена, который образуется вследствие t (9;22) —ABL-BCR на der (9), но роль его для развития хронического миелолейкоза не выяснена.
Делеция
Также установили очень важный факт: приблизительно 20-25 % пациентов, у которых обнаружен хронический миелолейкоз, имеют делецию в маркерной 9q+ хромосоме. Эту аномалию нельзя обнаружить при проведении простого хромосомного анализа, но можно увидеть ее во время использования FISH с зондами, которые специально разработаны.
Размер делеции варьируется у каждого пациента: участок с делецией может содержать последовательности BCR-гена, которые перенесены с 22-й хромосомы на 9-ю, на последовательности самой 9 хромосомы или обеих.
Установили также возникновение делеции с формированием специфической t (9;22) одновременно, что частота ее в группах пациентов, которых обследовали на различных стадиях хронического миелолейкоза, одинаковая.
Пока что применение данного метода диагностики (информативного прогностического), к сожалению, в широкую клиническую практику не вошло в связи с его сложностью и требованием дорогостоящего оборудования и реактивов.
Ее роль в прогрессии ХМЛ
Делеция маркера 9q+ играет важную роль в прогрессии ХМЛ, но до конца все еще не выяснена. Когда выпадают кодирующие последовательности генов ABL или/и BCR вследствие делеции, это приводит к экспрессированию лишь одного химерного гена BCR-ABL, но экспрессии гена ABL-BCR не происходит. Вероятно, данное событие также играет важную роль в прогрессировании лейкоза. Более того, исследователи обсуждают возможность инактивации пока еще неизвестных генов-супрессоров, которые локализованы в хромосомном делетируемом районе.
Белок с мол. м. 210 тысяч кодируется химерным геном BCR-ABL, который обладает большей, нежели продукт протоонкогена нормального ABL (H145), протеинкиназной активностью. Во время лейкоза, который у мышей вызван вирусом Абельсона, онкогенную активность имеет белок, продукт gag/abl гибридного гена, который имеет высокую протеинкиназную активность. В эксперименте проводилось вырезание gag/abl гена, после чего вирус уже не мог у мышей вызывать лейкоз. То есть филадельфийская хромосома является маркером этого заболевания.
Подробнее о генах BCR и ABL
При изучении в генах BCR и ABL разрывов при ХМЛ было выявлено, что у разных пациентов разная их локализация. К примеру, в ABL гене протяженность участка, где могут быть разрывы, большая, до 200 kb, а в BCR-гене локализация разрывов происходит на небольшом участке в 8,5kb, то есть можно говорить о наличии кластера разрывов, давшего название самому BCR гену - Breakpoint cluster region.
Во многих случаях t (9;22) разрывы BCR-гена обнаруживаются на участке M-BCR, причем в химерный ген включается длинная часть BCR гена, результатом чего становится появление белка, характерного для ХМЛ P210Bcr/Abl. К тому же при t (9;22) разрывы BCR-гена локализоваться могут на участках, которые называются m-bcr и u-bcr. Bm-bcr области разрывы ведут к образованию P190Bcr/Abl химерного белка, то есть меньшего по величине, чем Р210Bcr/Abl. А локализация разрывов в u-bcr ведет к образованию белка Р230Bcr/Abl более крупного.
Эти различия молекулярного характера коррелируют не строго с клинической особенностью лейкоза. Например, Р190Bcr/Abl имеется у 2 разных видов лейкозов: Ph-позитивный лимфобластный острый лейкоз и гранулоцитарный хронический лейкоз при выраженном моноцитозе и миелопластическими чертами. Если обнаруживают Р230Bcr/Abl, то можно наблюдать картину нейтрофильного хронического миелолейкоза, то есть миелолейкоз, формула крови при котором представлена единичными метамиелоцитами и зрелыми нейтрофилами. К тому же название нейтрофильного гранулоцитарного лейкоза долгое время используют, чтобы обозначать Ph(BCR-ABL)-негативный хронический миелолейкоз (относительно доброкачественный вариант, наблюдаемый у пожилых людей и иногда у подростков).
Любой из вышеприведенных белков можно обнаружить при хроническом миелолейкозе. Также сообщается о достаточно частом сочетании 2 типов белков (Р1900Bcr/Abl и Р210Bcr/Abl) при лимфобластном остром лейкозе и хроническом типичном миелолейкозе.
Роль BCR-ABL для развития хронического миелолейкоза
Решающую роль BCR-ABL гена и его продукта, а именно Р210 белка для развития хронического миелолейкоза демонстрировали на разных системах in vitro и in vivo. Например, при трансдукции bcr/abl в гемопоэтические стволовые клетки мышей и их дальнейшей трансплантации сингенным облученным животным у вторых возникает миелопролиферативная болезнь, похожая на хронический миелолейкоз человека. Установили, что онкогенный потенциал обуславливает высокая тирозинкиназная активность химерного белка BCR-ABL. Одним из центральных в злокачественной трансформации клеток является событие дерегуляции тирозинкиназной активности.
Когда исследователи вводили летально облученным мышам клетки, которые экспрессировали р210Bcr/Abl, то одни животные заболевали лейкозом, сходным с человеческим хроническим миелолейкозом, а другие различными новообразованиями из клеток гемопоэтических: эритроидные опухоли, миеломоноцитарные лейкозы, ретикулоклеточные саркомы, пре В- и Т-клеточные лимфомы, макрофагальные опухоли. Причину различий не выяснили. Такие опыты показывают, что всю цепь событий, приводящую к развитию хронического миелолейкоза еще не установили. Но факт остается. При этом заболевании обнаруживается филадельфийская хромосома в клетках костного мозга. Лечение рассмотрим ниже.
Что еще обнаружили у больных?
Получили данные, которые свидетельствуют о существенном увеличении массы кроветворных клеток и элементов крови в организме пациентов с хроническим миелолейкозом, в основном из-за резкого увеличения срока жизни таких клеток, потому что активированным геном ABL (в BCR-ABL гене) ингибируется апоптоз – запрограммированная клеточная смерть. К тому же этим геном усиливается пролиферация миелоидных клеток.
Имеются основания полагать, что в случае хронического миелолейкоза изменяется функция спец. клеточных белков, то есть интергинов, последствием чего становится нарушение адгезии к стромальным элементам молодых миелоидных клеток, а также происходит избегание стволовых лейкемических клеток негативных регуляторных влияний. Можно сделать вывод. Наличие филадельфийской хромосомы патогномонично для хронического миелолейкоза.
Выводы

Наиболее общей формулировкой молекулярного патогенеза является следующая: химерным геном BCR-ABL кодируется белок, у которого постоянно активирована тирозинкиназная активность, что приводит к активации большого количества сигнальных путей, а также выраженным изменениям апоптоза, адгезии и клеточного цикла. Считается, что эти события достаточны для определения злокачественной клеточной трансформации и поддерживания опухолевого фенотипа.
Но все равно основным маркером является филадельфийская хромосома.
Лечение
Для подавления активности патологических лейкоцитов необходимой для сохранения жизни больных им требуется вторая линия терапии. Для этого применяют нилотиниб – вещество, которое блокирует передачу сигнала от филадельфийской хромосомы.
Ведь именно это и заставляет костный мозг продуцировать в большом количестве поврежденные лейкоциты.
Специалистами выделяется 3 ступени. Во время первой достигается гематологическая ремиссия, нормализация анализа крови и состояния пациента (размера его селезенки).
Но в клетках все же остается филадельфийская хромосома. Во вторую очередь достигается цитогенетическая ремиссия, когда эта хромосома не определяется в клетках. В-третьих, при проведении молекулярного исследования клеток крови в результате воздействия ингибиторами тироксинкиназы у костного мозга не обнаруживают патологический ген.
Трансплантация костного мозга
Обязательна ли ТКМ при филадельфийской хромосоме? Такую операцию по трансплантации проводят больным с острой формой миелоидного лейкоза. Также реально совместимого донора найти бывает непросто. Это очень серьезная и длительная операция. Но она позволяет восстановить нормальную работу костного мозга.
Читайте также: