
Генетика рака схема механизмов развития
Рак — одна из наиболее частых и серьезных болезней, наблюдаемых в клинической медицине. Статистика показывает, что некоторые формы рака встречаются у более чем одной трети людей, вызывая более 20% всех смертей, и в развитых странах требуют более 10% общих расходов на медицинское обслуживание. Раковые опухоли при отсутствии лечения всегда приводят к летальному исходу.
Ранняя диагностика и раннее лечение жизненно необходимы, и немаловажная цель исследования рака — выявление людей с повышенным риском раковых опухолей до их развития.
В дальнейших статьях на нашем сайте МедУнивер попробуем разобраться, каким образом молекулярно-генетические исследования показывают, что рак — в основном генетическая болезнь. Во-первых, опишем типы генов, вовлеченных в развитие рака, и механизмы, благодаря которым дисфункция этих генов может заканчиваться болезнью.
Во-вторых, рассмотрим множество наследуемых онкологических синдромов и покажем, как понимание их патогенеза высветило основу более частых спорадических форм рака. Мы также изучим некоторые специальные проблемы, возникающие в медицинской генетике и генетическом консультировании в связи с наследуемыми синдромами.
В-третьих, покажем, как генетика и геномика изменили наши представления о причинах рака и методах его диагностики и лечения. Геномика за счет идентификации конкретных делеций и дупликаций сегментов генома раковых клеток и полного анализа экспрессии генов и мутаций в раковых клетках действительно изменила диагностику и лечение рака.
Рак — не одно заболевание, это название используют для обозначения злокачественных новообразований, характеризующихся неконтролируемым клеточным ростом, приводящим к их развитию. Новообразование, чтобы быть раком, должно также быть злокачественным.
Это означает, что его рост больше не контролируется, и опухоль способна прорастать смежные ткани или распространяться (метастазировать) в более отдаленные участки, или и то, и другое одновременно. Опухоли, не способные к прорастанию или метастазированию, не относятся к раковым и называются доброкачественными опухолями, хотя их размер и расположение могут вызывать беспокойство, но в целом они благоприятны для пациента.
Существует три основных формы злокачественных новоообразований: саркомы, когда опухоль возникает в мезенхимальной ткани, например в костях, мышцах, соединительной ткани или в тканях нервной системы; карциномы, возникающие в эпителиальной ткани, скажем, в эпителии клеток кишечника, бронхов или протоках грудной железы; и злокачественные неоплазии гемопоэтической и лимфоидной ткани, например лейкозы и лимфомы, захватывающие костный мозг, лимфатическую систему и периферическую кровь.
В пределах каждой из этих основных групп опухоли классифицируются по их расположению, типу ткани, гистологическим проявлениям и степени злокачественности.
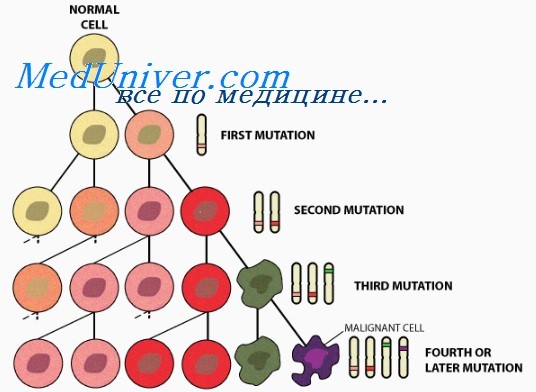
• Независимо от того, появился рак спорадически, в результате соматической мутации или у многих членов одной семьи как наследственный признак, это генетическое заболевание.
• Онкоген — мутантный аллель протоонкогена, класса нормальных генов, кодирующих белки клетки, обеспечивающие рост и выживание клеток. Онкогены облегчают злокачественное перерождение, стимулируя пролиферацию или тормозя апоптоз. Онкогены кодируют такие белки, как:
- белки сигнальных путей пролиферации клеток;
- факторы транскрипции, управляющие экспрессией обеспечивающих рост генов;
- ингибиторы механизмов программируемой смерти клетки.
• Развитие опухоли. После появления рак развивается, накапливая генетические поломки, благодаря мутациям или эпигенетическому подавлению генов ХКЦ, кодирующих механизмы репарации поврежденной ДНК и поддерживающих цитогенетически нормальное состояние. Другое последствие генетических дефектов — изменение экспрессии генов, приводящее к васкуляризации и распространению опухоли инвазивным ростом и метастазированием.

Александр Олегович Иванцов, доктор медицинских наук
— Чтобы ответить на этот вопрос, стоит разобраться как развивается опухоль. Она имеет автономный характер роста. Что это значит? В норме количество клеток в организме человека регулируется балансировкой двух противоположных процессов – клеточного деления и клеточной гибели. При росте опухоли прибавление клеточной массы опережает клеточную гибель. Это возможно по двум причинам – либо активируются процессы пролиферации, т.е. деления клетки, либо угнетается апоптоз, т.е. запрограммированная клеточная гибель. Автономность опухоли состоит в том, что ее клетки не способны реагировать на внешние сигналы организма, и, как следствие, она продолжает рост.
Если изменения нуклеотидной последовательности ДНК происходят в значащих фрагментах ДНК (прим. – экзонах), то они могут привести к развитию опухоли. К развитию рака приводят в основном мутации, нарушающие баланс деления и гибели клеток, то есть мутации в генах, контролирующих именно эти процессы. Мутации могут возникать случайно, например, в процессе удвоения ДНК в результате деления клетки. А могут возникать под влиянием мутагенов: например, воздействия ультрафиолетового или рентгеновского излучения, высокой температуры, некоторых химических веществ. На последний вопрос, можно ответить, что патогенность мутации можно предположить в первую очередь по функции гена, который она затрагивает, по её структурным характеристикам (насколько сильно она нарушает или изменяет работу этого гена), и подтвердить путем функциональных исследований (например, на клеточных культурах).
— Что такое онкогены?
— Онкогеном называется ген, который в норме не оказывает влияние на процессы деления и гибели клеток, а в опухоли активизируется, вследствие чего раковые клетки приобретают способность к неконтролируемому размножению. Кроме того, в настоящее время известно о роли антионкогенов. В норме они подавляют процесс деления клеток или способствуют их гибели, а в опухоли этот сдерживающий эффект подобных генов отсутствует, тем самым провоцируется рост опухолевых масс. Современная наука полагает, что для возникновения трансформированного клеточного клона необходимо как минимум пять-девять мутаций в разных онкогенах и антионкогенах.
— Эти мутации можно выявить с помощью генетического исследования?

— Кому и чем могут помочь генетические исследования? Верно ли, что от генетического исследования может зависеть успех лечения? Кому стоит пройти генетическое исследование на мутации?
— Сфера медицинского применения ДНК- и РНК-тестов в современной онкологии постоянно расширяется. Сейчас это тестирование позволяет диагностировать наследственные опухолевые синдромы, выявить предиктивные мутации, осуществить анализ экспрессионных характеристик опухоли. Также совершенствуются технологии, которые позволяют уточнять диагноз опухолей с невыявленным первичным очагом, эффективно контролировать течение заболевания и изменения свойств опухоли (жидкостная биопсия), выполнять различные биологические тесты с опухолевыми клетками.
Индивидуализация лечения онкологического пациента во многих случаях напрямую зависит от результатов генетического тестирования. Эмпирический подход, сопряжённый со случайным перебором биологически активных химикатов, постепенно замещается научно-обоснованным, молекулярно-направленным поиском специфических противоопухолевых средств, направленных на активацию или инактивацию ключевых биохимических компонентов опухолевой трансформации.
Например, еще недавно клиническое деление всех первичных опухолей легкого на мелкоклеточный и немелкоклеточный рак было достаточным для определения стратегии лечения. Ситуация изменилась с открытием активирующих мутаций в гене, который кодирует рецептор эпидермального фактора роста — EGFR, сделавших этот онкогенный белок избирательной мишенью для воздействия препаратов ингибиторов EGFR. Мутации EGFR, как правило, встречаются у пациентов с аденокарциномой легкого. Тест на мутацию EGFR позволяет практически со 100%-й достоверностью отобрать тех больных, у которых гарантирован положительный результат применения гефитиниба, эрлотиниба или афатиниба.
— Может ли генетическое исследование помочь здоровому человеку предупредить рак или выявить его на ранней стадии?
Генетическое исследование при подозрении на наследственный раковый синдром носит комплексный характер. Оно начинается со сбора онкологического анамнеза ‒ уделяется внимание случаям злокачественных заболеваний у кровных родственников. В результате составляются родословные, позволяющие заподозрить наследственную патологию. На заключительном этапе проводится анализ ДНК, что позволяет установить наличие в генотипе больного, а также членов его семьи, подозреваемые мутации.
— Какие виды мутаций ученые уже выявили? Существует ли для каждого вида таргетный препарат? Как именно работает таргетный препарат?
— Много разных видов мутаций при разных опухолях известны, но наибольший интерес представляют мутации в онкогенах, в частности, в рецепторных протеинкиназах, для блокировки которых разрабатываются специфические препараты. Мутации в протеинкиназах изменяют конформацию белковых молекул и, таким образом, формируют идеальное терапевтическое окно. Таргетный препарат избирательно воздействует на клетки опухоли, содержащие молекулярную мишень, и этим выгодно отличается от химиотерапии. Известно об успешном использовании ингибитора тирозинкиназы ALK – кризотиниба – у больных с ALK-транслоцированными карциномами легкого. Успешным оказалось и применение специфических ингибиторов мутированного белка BRAF – вемурафениба и дабрафениба для лечения больных меланомой. Другой пример: ген BRCA1 кодирует фермент репарации ДНК. BRCA1-дефицитные клетки демонстрируют неспособность эффективно удалять сшивки ДНК, индуцированные препаратами платины. В наследственных BRCA1-ассоциированных раках отмечается наибольшая эффективность цисплатина, т.к. в опухолевых клетках наблюдается соматическая утрата оставшегося BRCA1-аллеля, в то время как нормальные клетки носительниц мутаций BRCA1 сохраняют интактную копию данного гена. Этим обусловлено уникальное терапевтическое окно и это объясняет высокую эффективность цисплатина при лечении BRCA1-ассоциировнного рака молочной железы, яичника. Конечно, по разным причинам, не для всех мутаций есть такие препараты, но их спектр и количество неуклонно возрастает.
— В настоящее время проводятся исследования в двух направлениях: диагностика наследственных раковых синдромов и индивидуализация подбора лекарственных препаратов на основе молекулярных характеристик опухоли. Тем самым повышается клиническая эффективность применения дорогостоящих лекарственных препаратов, снижается частота и тяжесть побочных эффектов, и в некоторых случаях предотвращается неблагоприятный исход заболевания.
— В настоящее время проводятся исследования в двух направлениях – диагностика наследственных раковых синдромов и индивидуализация подбора лекарственных препаратов на основе молекулярных характеристик опухоли. Тем самым повышается клиническая эффективность применения дорогостоящих лекарственных препаратов, снижается частота и тяжесть побочных эффектов, и, в некоторых случаях, предотвращается неблагоприятный исход заболевания.
Новообразование — аномальное скопление клеток, вызванное дисбалансом между ростом и гибелью клеток. Клетки размножаются в ходе клеточного цикла и прохождения митоза. Программируемая смерть клеток удаляет их из ткани.
Развитие рака (онкогенез) вызвано мутациями в одном или более генов из обширного числа, регулирующих рост и программируемую смерть клеток. Когда рак развивается как часть наследственного ракового синдрома, мутация наследуется через половые клетки и, следовательно, присутствует в каждой клетке тела.
Большинство опухолей, тем не менее, спорадические, поскольку мутации происходят в единственной соматической клетке, затем делящейся и начинающей перерождаться в рак. Не удивительно, что соматические мутации могут вызывать рак. Чтобы получить из единственной зиготы взрослый организм с его предполагаемыми 10 14 клеток, требуется множество клеточных делений.
Если учесть данные частоты ошибок репликации (10 -10 на основание ДНК за одно клеточное деление) и предполагаемые 10 15 делений в течение всей жизни взрослого человека, только ошибки репликации в результате дают тысячи новых мутаций геномной ДНК в каждой клетке организма. К мутационному бремени добавляются геномные и хромосомные мутации. Гены, видоизменяющиеся при раке, в сущности, не более подвержены мутациям, чем другие гены.
Несомненно, в соматических клетках происходит множество мутаций, приводящих у одной из множества клеток к утрате ее функций или гибели, но не имеющих фенотипического эффекта, поскольку потеря одной клетки скрывается преобладающим большинством здоровых клеток в органе или ткани. Онкогенные мутации отличает то, что по своей природе они способны даже одну мутантную клетку преобразовать в угрожающую жизни болезнь.
Список генов, участвующих в онкогенезе, также включает гены, синтезирующие декодирующую РНК, из которой образуются регуляторные микроРНК (miRNAs). В геноме человека присутствует по крайней мере 250 типов интерферирующих микроРНК, выполняющих ингибирова-ние экспрессии целевых генов, кодирующих белки, или вызывая распад соответствующей мРНК, или блокируя ее транскрипцию.
Приблизительно 10% микроРНК оказались либо существенно избыточными, либо недостаточно экспрессирующимися в различных опухолях, иногда настолько резко, что получили название онкомикроРНК (oncomirs). Один из примеров — 100-кратное усиление экспрессии микроРНК miR-21 в многоформной глиобластоме, очень злокачественной форме рака мозга. Избыточная экспрессия некоторых видов микроРНК может подавлять экспрессию целевого гена-супрессора опухолевого роста, тогда как снижение функции других микроРНК может приводить к избыточной экспрессии регулируемых ими онкогенов.
Поскольку каждая интерферирующая микроРНК регулирует до 200 разных генов, избыток или недостаток экспрессии микроРНК может оказывать масштабный онкогенный эффект, поскольку нарушается регуляция большого числа генов.
Парадигма развития обеспечивает полезную концептуальную основу для изучения роли генетических нарушений при раке, рассматриваемой в настоящей главе. Это — общая модель, которая, вероятно, относится к множеству, если не к большинству онкологических заболеваний, хотя лучше всего это видно на примере рака кишечника.
Краткий очерк развития генетики рака
В настоящее время не вызывает сомнения тот факт, что рак - это генетическая болезнь, в основе которой лежат изменения в геноме клетки. То есть это болезнь, связанная с потерей, повреждением, активацией, или, наконец, привнесением извне определенных генов.
Развитие теории канцерогенеза проходило длительное время, в несколько этапов, и характеризовалось прогрессирующим усилением генетического паттерна. Развитие наиболее значимых концепций канцерогенеза – гормонально метаболической, иммунологической, вирусной и др., закономерно привело к пониманию того, что рак является, по существу, генетической болезнью. Именно генетическая методология позволила придать теоретическую форму представлениям об опухолевом росте. Хотя, несмотря на все успехи, полнота этих представлений весьма относительна.
Более полный вариант мутационной теории рака был сформулирован Бауером К.Х. в 1928 году, фактически на заре генетики (Bauer,1928) Согласно Бауеру, "не существует наследственной передачи рака в точном смысле этого выражения. … Речь идет о наследовании склонности тканей образовывать опухоли при определенных внешних условиях". Эта склонность появляется вследствие возникновения в тканях соматических мутаций, которые могут быть очень разнообразными и включают как генные, так и хромосомные изменения" (Горбунова, Имянитов, 2007). Несмотря на то, что в 1929 году было показано, что в раковых клетках значительно выше частота аномалий кариотипа, эти представления отошли на второй план и на десятки лет уступили место другим гипотезам.
В 70-е годы прошлого века было показано, что способность вируса к злокачественной трансформации определяется наличием всего одного гена, названного онкогена src. Позже установили, что аналогичный характер наследования характерен для большинства онкогенных вирусов.
Появилась возможность искать аналоги вирусных онкогенов в геномах других организмов. Оказалось, что они встречаются достаточно часто. В геноме человека присутствуют гомологи всех вирусных онкогенов. Дальнейшие исследования показали, что эти гомологи контролируют важнейшие этапы жизни клетки, такие как пролиферация, дифференцировка, клеточный цикл и др. В клетках человека присутствуют десятки онкогенов, не все из которых связаны с вирусными онкогенами. Безусловным достижением стало открытие факта активации онкогенов в опухолях. Оказалось также, что вирусные онкогены не являются точными копиями клеточных онкогенов. Они несут многочисленные мутации, в том числе и довольно протяженные хромосомные перестройки. Именно эти изменения определяют трансформирующую способность вирусных онкогенов.
Таким образом, в результате разработки метода гибридизации нуклеиновых кислот было открыто, что все вирусные онкогены имеют гомологи в составе генома человека. Они ответственны за пролиферацию, дифференцировку, контроль клеточного цикла.
Доказательство определяющей роли мутагенеза в формировании онкогенности стало важной вехой мутационной теории рака. Именно эти представления послужили основой открытия антионкогенов. В 80-е годы XX века было установлено, что практически каждая опухоль, кроме мутаций в онкогенах, содержит множественные мутации в так называемых антионкогенах. Эти мутации могут быть как точковыми, так и делеционной природы. Предполагается, что инактивирующие мутации в генах супрессирующих развитие опухолей встречаются гораздо чаще, чем доминантные мутации, активирующие онкогены.
Открытие протоонкогенов и генов-супрессоров послужило прямым доказательством мутационной природы рака. Было показано, что изменение их структуры и экспрессии за счет различных мутационных процессов приводит к злокачественной трансформации (Rayter et al., 1989).
Благодаря развитию молекулярной медицины в 90-х гг. было доказано, что рак относится к генетическим мультифакторным заболеваниям. Было накоплено огромное количество фактов, которые свидетельствовали о мутационной природе рака. Прежде всего, это мутагенность практически всех известных физических и химических канцерогенов (Ауэрбах, 1978).
Был осуществлен перенос трансформированного фенотипа с помощью ДНК от злокачественных клеток (спонтанно и химически трансформированных) и опухолей в нормальные (Perucho et al., 1981; Shin et al., 1979). Существование тесной взаимосвязи темпов спонтанного возникновения генных мутаций и отдельных признаков злокачественнной трансформации стало доказательством мутационного происхождения последних (Varshaver et al., 1983; Bellett et al., 1979).
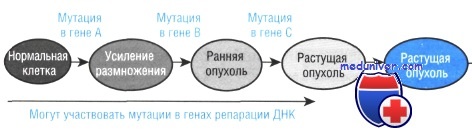
К началу 90-х гг. в онкологии сложилась точка зрения, согласно которой причиной возникновения большинства опухолей считается сочетание мутаций в онкогенах и антионкогенах. Как полагает современная наука, минимум 5-9 мутаций необходимо в различных онкогенах и антионкогенах, чтобы возник трансформированный клеточный клон (Kinzler, Vogelstein, 1997). При частоте мутирования, равной 10-6-10-5, характерной для нормальных соматических клеток, злокачественная трансформация была бы практически невероятным событием. Очевидно, чаще всего подобные мутации возникают последовательно и вне зависимости друг от друга. Это предопределяет путь развития заболевания и приводит к завершению процесса начавшейся неоплазии. По всей видимости, на каком-то этапе трансформации опухолевый клон приобретает свойство геномной нестабильности, т.е. способность к ускоренному мутагенезу. При этом с каждой новой мутацией происходит как бы последовательное "клонирование" трансформированных клеток с наращиванием потенциала их канцерогенности.
Проверка достаточно логичного предположения о том, что на каком-то этапе трансформации опухолевый клон приобретает гипермутабильный фенотип, стала одним из основных направлений молекулярной генетики рака в 90-е годы прошлого века. Исследования подтвердили существование генетической нестабильности опухолей - этот феномен продолжает интенсивно изучаться и в настоящее время.
Таким образом, во многом благодаря успехам генетики, молекулярная онкология вошла в XXI век вооруженная достаточно четкими представлениями о патогенезе новообразований. Молекулярно-генетические изменения, ведущие к развитию неоплазм, сводятся к трем последовательно реализующимся компонентам: 1) активация онкогенов; 2) инактивация опухолевых супрессоров; 3) развитие геномной нестабильности. Такое развитие событий определяет характерное для опухолей разнообразие генетических повреждений.
Семейные случаи рака
В большинстве случаев хромосомные мутации возникают в одной клетке спонтанно, под влиянием факторов внешней среды. Дальнейшее бесконтрольное деление этой клетки приводит к возникновению раковой опухоли. Случаи такого рака называются спорадическими, они не связаны с наследственностью. Спорадические формы рака встречаются с существенно большей частотой.
Известно, что некоторые формы злокачественных опухолей могут изредка возникать у нескольких членов одной семьи. Это касается и раковых опухолей плотных тканей и опухолевых заболеваний кроветворной системы. Когда измененные хромосомы унаследованы при рождении и присутствуют в каждой клетке тела человека, такая форма рака называется наследственной.
Семейная форма рака обнаруживается у членов одной семьи и вероятность возникновения заболевания у здоровых членов семьи в несколько раз выше, чем в случае спорадического рака. Заболевание показывает четкую наследственную картину.
Заранее узнать, кто из родственников – носитель мутантных генов, невозможно, - только половина родственников являются носителями мутаций того или иного гена. Все члены таких семей составляют группу повышенного риска. Ранее "семейную онкологию" связывали с многочисленными факторами, включающими канцерогены, ионизирующее облучение, экологические воздействия или вирусы, вызывающими опухоли. Однако научно-экспериментального подтверждения теории, связанные с этими факторами, не взирая на длительное и кропотливое обследование членов таких семей, не получили.
Достижения молекулярной биологии привели к открытию наследственных генетических дефектов, которые приводят к развитию рака.
К наследственным опухолевым синдромам относятся те заболевания, которые характеризуются 80-100% риском появления новообразований. К характерным особенностям наследственных злокачественных опухолей относится вероятность их возникновения в раннем периоде онтогенетического развития. Семейные случаи заболевания раком составляют всего лишь несколько процентов от всех пациентов онкологических клиник.
Наследственные опухолевые синдромы можно условно отнести к двум группам. Это редкие болезни, при которых опухоли не являются главными клиническими проявлениями; и "семейные раки" с довольно высокой встречаемостью. При этом развитие опухолей в каждом случае идет одинаковым путем и обусловлено наследованием гетерозиготных инактивирующих мутаций в супрессорных генах.
Семейные случаи злокачественных заболеваний однородны по составу мутаций, повреждающих гены-супрессоры. В связи с этим они легко поддаются ранней диагностике и даже профилактическому лечению.
Каковы же механизмы передачи предрасположенности к определенному виду опухолей? Все дело в существовании рецессивных онкогенов. Если в соматической клетке поврежден один аллель подобного гена, клетка остается фенотипически нормальной. Если же произошла мутация и в материнской и в отцовской копии, клетка приобретает черты злокачественной трансформации. Что произойдет, если мутация одного из аллелей рецессивного онкогена передастся пациенту через гаметы? Все соматические клетки будут иметь лишь одну "здоровую" копию. Повреждение оставшегося интактного аллеля всего лишь в одной из миллионов клеток органа-мишени приведет к возникновению клона с потенциями к злокачественному росту.
Важно то, что если на уровне клетки подобные нарушения носят рецессивный характер, то на уровне организма наследование происходит по доминантному типу. Носительство зародышевых раковых мутаций бывает лишь гетерозиготным. По всей видимости, аналогичные гомозиготные состояния и нормальное развитие эмбриона попросту несовместимы (Имянитов и др., 1993, Имянитов, Хансон, 2007; Caldas, Ponder, 1997).
Таким образом, из вышесказанного можно вывести клинико-генетические характеристики наследственных опухолевых синдромов:
- доминантный тип наследования (гетерозиготы, то есть лица с врожденным поражением только одного из двух аллелей антионкогена, являются "больными");
- высокая встречаемость онкологической патологии среди кровных родственников больного;
- необычно ранний возраст появления неоплазм (вполне достаточно мутации в одном аллеле антионкогена, а не в двух; в результате чего функциональная инактивация последнего у носителей происходит намного быстрее, чем у здоровых);
- множественность опухолей (т.е. вероятнее возможность того, что блокировка супрессорного гена произойдет в двух независимых клонах (Quesnel, Malkin, 1997; Caldas, Ponder, 1997; Имянитов, Хансон, 2007).
В настоящее время все основные типы семейных раков и соответствующие им мутации идентифицированы.
Таким образом, как мы уже упоминали выше, рак - заболевание с ярко выраженной наследственной компонентой. Тем не менее, это не значит, что заболеет каждый, у кого имеется онкологически больной родственник. Целый ряд генов-супрессоров опухолевого роста препятствует неконтролируемому клеточному делению, и направляет сильно поврежденные клетки на путь апоптоза - запрограммированной клеточной смерти.
Недавно было установлено, что предрасположенность к возникновению семейных раковых опухолей создается специфическими изменениями гена, расположенного в 13-й хромосоме, который имеет свойства супрессора опухолей.
Например, при некоторых видах рака наблюдается мутация в 13-й хромосоме, в которой находится ген ARLTS1, который, как предположили, участвует в супрессии роста опухолей. Calin с соавт. (2005) исследовали участок 13-й хромосомы, содержащей данный ген. Анализу подверглись 216 образцов ДНК клеток спорадических опухолей лёгких, щитовидной железы, кишечника, молочных желёз, а также кровь 109 пациентов с семейными опухолями различной локализации. Контролем служили образцы крови 475 здоровых людей или больных с заболеваниями, неопухолевой природы
Анализ образцов ДНК опухолей и клеток крови онкологических больных показал, что во всех видах злокачественных опухолей ген ARLTS1 заторможен.
Дополнительное экспериментальное исследование на животных позволило установить, что искусственное блокирование гена ARLTS1 действительно способствует активному появлению и развитию опухолей в различных тканях организма.
Таким образом, было установлено, что ген ARLTS1 в 13-й хромосоме имеет свойства супрессора опухолей. Специфические изменения в нём предрасполагают к возникновению семейных раковых опухолей.
Спорадическая форма рака также имеет генетическую компоненту, но она намного сложнее. Опухоль в этом случае развивается под воздействием не одной, а целой группы мутаций. Исследования полиморфизма генов, вовлеченных в канцерогенез, позволили выявить такие сочетания мутантных аллелей генов, которые существенно повышают риск возникновения рака. Спорадические опухоли не так просто обнаружить. Для большинства таких опухолей не найдено никакого генетического изменения, однозначно указывающего на то, что процесс злокачественного роста запущен. Генетическая сложность в этих случаях не позволяет провести раннюю диагностику, либо подобрать специфическое лечение.
Благодаря комбинационной изменчивости такие сложные генотипы распадаются уже в следующем поколении. Этим и объясняется отсутствие больных родственников у пациентов со спорадическим раком.
В случае подозрения наследственного опухолевого синдрома проводят генетическое исследование. Составляют родословные, которые помогают выявить наследственные патологию. Анализ ДНК позволяет установить, имеются ли в генотипе больного и его родственников подозреваемые мутации. Выявление и диагностика наследственных раковых болезней – весьма сложный процесс. К тому же полученная информация приносит свои положительные результаты только в том случае, когда уже имеются эффективные меры профилактики и лечения выявленных новообразований.
Мир семимильными шагами двигается к персонифицированной медицине, где важнейшую роль будет играть изучение генетических факторов человека. Это знание поможет не только вылечивать, но и предотвращать страшные болезни, например рак.
Анджелина Джоли – не просто голливудская звезда, но и настоящий боец! Мать и тётя актрисы в относительно молодом возрасте погибли от так называемого наследственного опухолевого синдрома. И Анджелина со свойственной ей решительностью превентивно (то есть заранее) удалила себе молочные железы и яичники (органы – мишени для наследственного рака, высокий риск которого обнаружил её генетический анализ). Первой из публичных личностей она открыто призналась, что сделала операцию, чтобы спастись от ещё несуществующей, но без экстренных мер практически неотвратимой угрозы. Личный риск Джоли, по подсчётам врачей, составлял 87%. Так она создала важный прецедент, который наверняка поможет сохранить жизнь многим женщинам во всём мире!

Это не случайность
Спорадический (то есть случайный) рак может возникнуть у любого. Главная причина болезни – генетические мутации в клетках. С годами в организме их накапливается критическое количество, поэтому развитие опухолей в пожилом возрасте – явление закономерное и почти неотвратимое. Как любят говорить онкологи, каждый человек доживёт до своего рака, если только раньше не погибнет от инфаркта или инсульта.
Но есть и наследственные формы болезни, поколениями поражающие людей из одной семьи. Путь к раку для носителей определённых мутаций в генах короче, чем у других людей. Если обычной клетке для превращения в раковую необходимо накопить 5–6 значимых мутаций, то при наследственном генном дефекте достаточно 4–5.
На долю наследственных опухолевых синдромов приходится лишь 5% среди всех онкологических заболеваний. Но риск стать жертвой страшной болезни для людей, которые носят в себе дефектный ген, не просто выше, чем у других, – он практически фатален. Например, риск заболеть раком молочной железы для обычной женщины составляет около 8–12%, а для носительниц мутаций в гене BRCA1 – больше 60%.
Тревожные признаки
Причина наследственного рака – в полученной от родителей мутации какого-то одного гена, из-за которого опухоль развивается в конкретном органе. Причём наличие семейной истории в этом случае не так важно. Например, если брать самый частый из наследственных синдромов рака молочной железы (РМЖ) и яичников (РЯ), то он может проявиться даже у женщины, в семье которой никто не страдал этим заболеванием. Ведь патогенную мутацию можно унаследовать и от отца.
У наследственного опухолевого синдрома есть чёткие клинические признаки:
Вид наследственного рака
Вклад
в заболеваемость
Гены, мутация в которых повышает риск
Рак молочной железы
BRCA1, BRCA2, CHEK2 и другие
Рак толстой кишки
MLH1, MSH2, MSH6, PMS2
CDH1, BRCA1, BRCA2
Рак поджелудочной железы
Рак предстательной железы
Медуллярный рак щитовидной железы
Приговор отменяется!
Например, при угрозе наследственного рака молочной железы и яичников мероприятия по ранней диагностике (УЗИ, маммография, сдача онкомаркеров) не слишком эффективны. Доказано, что они не гарантируют своевременное выявление опухоли. А рак яичников и вовсе часто выявляется только на запущенных стадиях. Более того, целый ряд новообразований склонен давать метастазы и на первой стадии, поэтому даже раннее выявление заболевания не всегда спасает от смерти. Особенно это характерно для некоторых наследственных опухолевых синдромов. Так, при выявлении РМЖ на первой стадии шансы прожить 5 и более лет есть как минимум у 98% женщин, а для носительниц мутаций в гене BRCA1 этот показатель всего 82%.

Наследственный медуллярный рак щитовидной железы относится к редким заболеваниям и вызывается патогенными мутациями в онкогене RET. Выявление у ещё здорового человека такой мутации – веское основание, чтобы заранее удалить щитовидку. Установлено, что просто частые наблюдения не защитят человека от гибели, а своевременная операция практически полностью снижает риск заболевания. Утрата органа эффективно компенсируется заместительной гормонотерапией.
При угрозе наследственного рака толстой кишки регулярные наблюдения, наоборот, очень эффективны. Исследования доказали, что, если делать колоноскопию каждые 1–2 года, можно достоверно уменьшить риск смерти от этой болезни. Профилактические операции на здоровой толстой кишке обычно не проводятся. Как не удаляют и те органы, утрата которых ведёт к существенному снижению качества жизни. Поэтому органы желудочно-кишечного тракта, части скелета, головной мозг хирурги, конечно, удалять не станут.
Важно
Одно из самых значимых достижений биомедицины последних лет – полногеномное секвенирование (NGS). Оно позволяет всего за несколько дней проанализировать ДНК любого человека. Благодаря ему наши знания о генетических патологиях многократно увеличатся уже в ближайшем будущем. Возможно, полногеномный анализ, который пока ещё слишком дорогое удовольствие (стоит тысячи долларов), в скором времени станет инструментом скрининга.
1. Делайте прививки!
Прививка от гепатита В (вызывает рак печени) и ВПЧ (причина рака шейки матки) может предотвратить 1,1 млн случаев рака в год.
2. Прогресс в онкогематологии
В конце ХХ века 60% детей, больных острым лейкозом, умирали, а сейчас в 90% случаев это заболевание излечивается полностью.
3. Не опускайте руки!
Сейчас в мире насчитывается около 28 млн человек, излечившихся от рака. Большинство из них – женщины, победившие рак груди.
Читайте также: