
Лечение хронического миелолейкоза гливек
С начала 1990 г., когда в результате разностороннего изучения патогенеза хронического миелоидного лейкоза (ХМЛ) стало очевидным, что обусловленная t(9;22)(q34;q11) активация ABL-тирозинкиназы является пусковым механизмом развития болезни, стали разрабатывать препарат, обладающий ингибирующим тирозинкиназу эффектом.
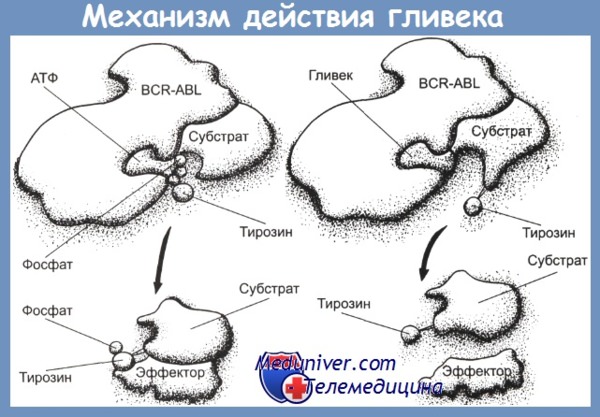
Гливек является производным 2-фениламинопиримидина (CH2SО3H). Исследования в культуре клеток больных ХМЛ показали, что гливек подавляет рост Ph-позитивных клеток, почти не действуя на Ph-негативные клетки. Механизм блокирующего активность ABL-тирозинкиназы действия гливека связан с встраиванием молекулы препарата в то место в молекуле ABL-тирозинкиназы — АТФ-карман, куда обычно встраивается АТФ.
С помощью рентгеновской кристаллографии доказано, что гливек встраивается в этот карман вместо АТФ, препятствуя тем самым фосфорилированию тирозина. Это приводит к блоку сигнала пролиферации в BCR-ABL-позитивных клетках, в результате чего они подвергаются апоптозу. В культуре клеток К562 показано очень быстрое действие гливека: фосфорилирование прекращается уже через 1 мин после введения гливека, подавление пролиферации и апоптоз — после 16—20 ч инкубации. В то же время исследования на больных показали необходимость значительно более продолжительного воздействия гливека: подавление фосфорилирования достигалось лишь через 3—4 дня.
Гливек принимают внутрь. Биодоступность гливека при пероральном приеме составляет более 97 %, она не зависит от возраста и приема пищи. Фармакологические исследования не обнаружили влияния массы тела на концентрацию гливека в плазме, поэтому препарат дозируется независимо от этого показателя. Для гливека характерна быстрая всасываемость с максимумом концентрации в плазме через 1—2 ч после приема. Время полувыведения препарата составляет от 18 до 22 ч, что позволяет принимать его один раз в день.
Примерно 96 % препарата связывается с белками плазмы, особенно с альбумином и гликопротеинами и в очень незначительной степени — с липопротеинами. Более 80 % гливека и его метаболитов выводится в течение 7 дней после приема однократной дозы. Препарат выводится главным образом (68 %) через кишечник, и только около 13% — почками.
По всей вероятности, гливек не проникает через гематоэнцефалический барьер. В опытах на животных показано, что даже при назначении очень высоких доз гливека, в результате чего концентрация в плазме в 10 раз превышает терапевтическую, в цереброспинальной жидкости она в 3 раза ниже терапевтической и в 155 раз ниже, чем в плазме.
В отечественных исследованиях получены столь же высокие результаты. По данным Гематологического научного центра РАМН, полный гематологический ответ при лечении гливеком получен у 96 % больных в хронической стадии хронического миелоидного лейкоза, ранее лечившихся ИНФ-а в комплексе с Ara-C, у 43 % больных — большой цитогенетический ответ, при этом в течение 18 мес лечения частота полной цитогенетической ремиссии увеличилась с 16 до 37 %.
Даже в фазе акселерации через 6 мес лечения у 30 % больных получен большой цитогенетический ответ, а при продолжении лечения до 18 и 24 мес — у 45 и 51 % соответственно при 39 % полного цитогенетического ответа.
Ухудшение гематологических или цитогенетических показателей за время лечения наблюдали у 10,3 % больных, получавших ИФН-а + Ara-C, и лишь у 1,4 % леченных гливеком, переход в стадию акселерации или развитие бластного криза — у 4,7 и 1,1 % соответственно. Немаловажной оказалась и разница в переносимости терапии: перевести в противоположную группу из-за непереносимости лечения пришлось 19 % больных из группы леченных ИФН-а + + Ara-C и лишь менее 1 % больных, получавших гливек.
В 2003 г. были опубликованы результаты этого исследования после 19 мес лечения. Полная гематологическая ремиссия достигнута у 97 % получавших гливек и 69 % леченных ИФН-а + Ara-C. Большой цитогенетический ответ достигнут и сохранился в течение всего срока наблюдения у 87 % больных, получавших гливек, и лишь у 35 % леченных комбинацией ИФН-а и Ara-C, a полная цитогенетическая ремиссия — соответственно у 76 и 14 %. Результаты этого исследования показывают, что при терапии гливеком, проводимой с самого начала заболевания, большинство больных хроническим миелоидным лейкозом имеют шанс прожить 10 лет.
Числовая сравнительная оценка качества жизни показала статистически достоверный более высокий уровень в группе получавших гливек. Более того, в группе больных, вначале получавших комбинацию ИФН-а и Ara-C, a затем переведенных на лечение гливеком, общая оценка качества жизни за время наблюдения оказалась выше, чем у больных, продолживших терапию ИФН-а + Ara-С.
Анализ течения болезни у 553 больных, с самого начала получавших гливек, провели после 2 лет терапии. Данные анализа показали следующее: полная гематологическая ремиссия сохранилась у 95 % больных, большой цитогенетический ответ констатирован к концу срока наблюдения у 88 %, полная цитогенетическая ремиссия — у 79 %, не было перехода в более продвинутую стадию у 96 %, выживаемость также составила 96 %. В зависимости от прогностического индекса по Socal оказалось, что полная цитогенетическая ремиссия достигнута и сохранялась в течение анализируемого срока у 84 % больных группы низкого риска прогрессирования, у 77 % группы промежуточного и у 62 % больных группы высокого риска. Течение заболевания без признаков прогрессирования отмечено соответственно у 95, 89 и 85 % больных. Ни при каком лечении до появления гливека не было подобных результатов.
Недавно подведенные итоги 5-летних наблюдений за больными, вошедшими в это исследование, показали, что терапия гливеком принципиально изменила течение хронического миелоидного лейкоза. Через 5 лет после начала терапии гливеком ранее не леченных больных ХМЛ в хронической стадии полная гематологическая ремиссия сохранялась у 97 % больных, большой цитогенетический ответ наблюдали у 88 %, полная цитогенетическая ремиссия была у 82 %. Общая 5-летняя выживаемость составила 90 %, у 93 % больных сохранялась хроническая стадия заболевания.
Менее чем у 1 % больных ежегодно наблюдалось прогрессирование в стадию акселерации или развитие бластного криза. Не отмечали прогрессирования болезни у 96 % больных, у которых в течение первых 12 мес лечения была достигнута молекулярная ремиссия (уменьшение транскрипта BCR/ABL более чем на 3 log; 3 log — это в 1000 раз), и у 81 % больных, у которых наблюдалась полная цитогенетическая ремиссия, но не было достигнуто молекулярной ремиссии. Как известно, при терапии миелосаном, начиная со второго года наблюдения, переход в стадию акселерации или развитие бластного криза ежегодно наблюдали у 15—20 % больных.
В 2004 г. опубликованы результаты лечения гливеком 235 больных в стадии акселерации и 260 больных в стадии бластного криза. Больные получали 600 мг препарата ежедневно. Через 36 мес от начала лечения были живы 55 % больных, начавших лечение в стадии акселерации и 14 % в стадии бластного криза. Выживаемость строго коррелировала с достижением большого цитогенетического ответа: 3-летняя выживаемость больных в стадии акселерации при достижении такого ответа в течение первых 3 мес лечения составила 85 % и 52 % больных без большого цитогенетического ответа.
Результаты лечения больных в поздней хронической стадии при лечебных дозах препарата 400—600 мг/сут были еще лучше: большой цитогенетический ответ получен у 65% больных, полный — у 52%. У 82 % больных большой цитогенетический ответ сохранялся в течение 3 лет.
Живы в течение 3 лет 88 % больных этой группы, причем у 80 % из них без признаков прогрессирования болезни.
Наилучшая выживаемость достигается у больных, у которых большой цитогенетический ответ в хронической стадии или любой цитогенетический ответ в стадии акселерации получен в течение первых 3 мес терапии. В стадии акселерации получение цитогенетического ответа после 3 мес лечения гливеком оказалось даже более значимым прогностическим фактором, чем наличие клональной эволюции. При получении даже минимального цитогенетического ответа в течение первых 3 мес у 83 % больных позже достигается большой цитогенетический ответ, при этом у 54 % — полная цитогенетическая ремиссия.
Если минимальный цитогенетический ответ получен в течение 6—12 мес терапии, то частота большого цитогенетического ответа и полной цитогенетиче-ской ремиссии в дальнейшем составляли соответственно 68 и 35 %. В тех случаях, когда в течение первых 3 мес лечения не наблюдали даже минимального цитогенетического ответа, только у 12 % больных можно было ожидать его достижения в дальнейшем, а при отсутствии цитогенетического ответа в первые 6 мес — только у 4 % больных. При неполучении цитогенетического ответа в течение 12 мес лечения возможность его достижения в дальнейшем равна 0.
Исследование экспрессии 380 генов у ранее не леченных больных, получавших гливек в течение 12 мес, позволило выявить 18 генов, по уровню экспрессии которых различались больные с молекулярной ремиссией к этому времени и те, у кого молекулярного ответа получено не было. Оказалось, что у резистентных к гливеку в 1,9—7,5 раза более выражена экспрессия генов, участвующих в передаче пролиферативного сигнала, процессах апоптоза, клеточной адгезии и регуляции клеточного цикла.
Накопление опыта в лечении гливеком показало, что стабильная гематологическая и цитогенетическая ремиссия сохраняется в тех случаях, когда удается добиться уменьшения количества BCR-ABL-позитивных клеток не менее чем на 3 логарифма (3 log). Уменьшение на 3 log и более количества определяемого транскрипта BCR-ABL (по соотношению BCR-ABL/BCR) рассматривается как большая молекулярная ремиссия. Анализ данных, полученных после 30 мес лечения, показал отсутствие про-грессирования заболевания у всех (100%) больных, у которых в течение первые 12 мес лечения достигнута редукция патологического клона на 3 log или более, и у 93 % больных с редукцией этого клона менее чем на 3 log. Течение болезни без прогрессирования в период, равный 30 мес, наблюдали у 82 % больных, у которых не было получено полной цитогенетической ремиссии в первые 12 мес лечения.
Показано, что при назначении гливека по 600—800 мг/сут и одновременном проведении двух 7-дневных курсов терапии цитозин-арабинозидом по 200 мг/м2 в сутки через 18 мес лечения у 51 % больных была достигнута редукция опухолевого клона более чем 3 log, а у 28 % — более чем на 4,5 log. Из 127 больных, получивших такое лечение, через 18 мес были живы 99 % и 94 % не имели признаков прогрессирования болезни. Лишь у 30 больных не была достигнута большая молекулярная ремиссия (не удалось уменьшить количество BCR-ABL-позитивных клеток на 3 log и более). У двух из них обнаружены мутации в ABL-тирозинкиназном домене белка BCR-ABL. Эти данные подтверждены в кооперированном исследовании из Италии, в котором участвовало 23 центра.
Больным, которые по своим клинико-гематологическим показателям относились к группе промежуточного прогноза по системе Socal, в качестве первоначального лечения назначали 800 мг гливека в сутки. Через 6 мес лечения полная гематологическая ремиссия достигнута у 98 %, полная цитогенетическая — у 87 % больных. После 12 мес лечения полная цитогенетическая ремиссия констатирована у 94 % больных, большая молекулярная ремиссия (уменьшение транскрипта BCL/ABL более чем на 3 log) — у 56 %.
Гливек большинством больных переносится очень хорошо. Наиболее частые проявления негематологической токсичности (тошнота, рвота, диарея, головные боли, мышечные судороги, отеки) возникают у 30—50 % больных, но только у 1—2 % их выраженность превышает I—II степень по шкале ВОЗ и требует временной отмены препарата.
Обычно суточную дозу гливека 400 мг назначают однократно натощак или через 1,5—2 ч после завтрака. Однако у ряда больных при приеме препарата натощак может наблюдаться нерезко выраженная тошнота. В таких случаях рекомендуется принимать препарат вместе с пищей, а если тошнота сохраняется и после этого, разделить дозу препарата на 2 приема.
-->| Радиоперехваты круглых столов [8] |
| Радиомагазин-73 [505] |
Войти через uID
Заболевания, входящие в группу ХМПЗ, возникают в результате злокачественной трансформации полипотентной гемопоэтической стволовой клетки костного мозга и последующей клональной пролиферации клеток одной или нескольких линий миелопоэза, сохраняющих способность к дифференцировке.
По классификации ВОЗ выделяют группу истинных ХМПЗ и группу миелопролиферативных/миелодиспластических заболеваний (МПЗ/МДЗ).
К ХМПЗ относятся:
1. Хронический миелолейкоз (bcr/abl положительный)
2. Хронический нейтрофильный лейкоз
3. Хронический эозинофильный лейкоз/гиперэозинофильный синдром
4. Хронический идиопатический миелофиброз
5. Истинная полицитемия
6. Эссенциальная тромбоцитемия
7. Миелопролиферативное заболевание, неклассифицируемое.
В группу МПЗ/МДЗ входят:
1. Хронический миеломоноцитарный лейкоз
2. Атипическии хронический миелоидный лейкоз
3. Ювенильный миеломоноцитарный лейкоз.
В МКБ-10 ХМПЗ рассматривается в группе опухолевых заболеваний:
D45 — Полицитемия истинная;
D47.3 — Эссенциальная (геморрагическая) тромбоцитемия;
С92.1 — Хронический миелоидный лейкоз. Идиопатический миелофиброз.
ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ
Эпидемиология. На долю ХМЛ приходится 15-20% всех случаев лейкозов у взрослых и 5% — у детей.
Заболеваемость ХМЛ составляет 15 новых случаев на 1 млн населения в год. Заболевание встречается преимущественно у лиц 30—70 лет, пик заболеваемости — 30—50 лет.
Мужчины и женщины страдают ХМЛ одинаково часто.
Этиология неизвестна, как и для всех опухолей.
К возможным этиологическим факторам относят малые дозы ионизирующего излучения, ряд химических веществ.
Патогенез. Пусковым моментом в развитии ХМЛ является соматическая мутация плюрипотентной гемопоэтической стволовой клетки крови. Основу мутации составляет перекрестная транслокация хромосомного материала между 9-й и 22-й хромосомами с образованием Ph-хромосомы и химерного онкогена bcr/abl на 22 хромосоме.
В отдельных случаях (частота их не превышает 5%) при стандартном цитогенетическом исследовании не удается обнаружить Ph-хромосому, в то время как молекулярно-генетическое исследование выявляет наличие онкогена bcr/abl.
Продуктом данного химерного гена при ХМЛ является белок р-210, являющийся тирозинкиназой с повышенной активностью, в результате чего нарушается нормальное функционирования клетки н ее злокачественная трансформация, бесконтрольная пролиферация гемопоэтических клеток.
В течении ХМЛ выделяют три фазы:
Хроническая (развернутая) — характеризуется пролиферацией клеток миелоидного ростка (гранулоцитарного, мегакарноцитарного) с сохраненной дифференцировкой клеток.
Стадия акселерации — характеризуется развитием резистентности к проводимой терапии и поя&чением нового клона злокачественных клеток с блоком дифференцировки на уровне бластных клеток.
В основе пояаления нового клона лежат вторичные мутации в опухолевых клетках.
Нарушение созревания приводит к увеличению в костном мозге и периферической крови количества незрелых клеток — бластов и промиелоцитов, бластный криз — характеризуется преобладанием клона с блоком дифференцировки над клоном клеток bcr/abl.
Костный мозг представлен большим количеством бластных клеток (эти клетки могут нести на своей цитоплазматической мембране маркеры, указывающие на их принадлежность как к миелоидному, так и к лимфоидному ростку).
Количество эритроцитов и НЬ в большинстве случаев находится в пределах нормы или несколько выше нее.
Количество тромбоцитов нормальное или повышенное. Все клетки крови имеют Ph-хромосому и ген bcr/abl.
Фаза акселерации (6—8 мес): появляются признаки прогрессирования лейкоза: лихорадка, боли в костях, нарастающая спленомегалия, у 25% больных — увеличение лимфатических узлов.
Гемограмма: лейкоцитоз (50—500)х10*9/л. Количество бластных клеток в периферической крови или костном мозге от 10 до 19% (по данным некоторых авторов — до 29%), количество бластов и промиелоцитов более 30%, характерны базофилия более 20%, нормохромная или гиперхромная анемия, персистирующий тромбоцитоз или тромбоцитопения. не связанная с терапией.
Появляются дополнительные хромосомные мутации (дополнительная Ph-хромосома, трисомия 8, изохромосома 17 и др.).
Бластный криз (средняя продолжительность фазы 3-6 мес): анемия, тромбоцитопения, признаки экстрамедулярных очагов кроветворения. Нарастают проявления геморрагического диатеза петехиально-пятнистого типа, связанного с тромбоцитопенией.
Характерны лихорадка, упорные боли в костях, быстропрогрессирующее истощение, быстрое увеличение селезенки и печени.
Гемограмма и миелограмма: количество бластов в костном мозге и периферической крови более 30%.
Диагностика. Морфологическое исследование крови и костного мозга подтверждает наличие миелопролиферативного процесса.
Диагноз ХМЛ подтверждается цитогенетическим исследованием, выявляющим наличие филадельфийской хромосомы и гена bcr/abl.
Разрешающая способность стандартного цитогенетического исследования — 5%, метода флюоресцентной гибридизации in situ (FISH) — 1 лейкемическая клетка на 200-500 нормальных.
Полимеразная цепная реакция используется как для диагностики, так и для мониторинга остаточной минимальной болезни.
Дифференциальная диагностика.
1. С лейкемоидными реакциями по нейтрофильному типу (нейтрофилы более 7,5x10*9/л — острые и хронические инфекции, неинфекционные хронические заболевания, ацидозы различной природы, на фоне терапии кортикостероидами, хронические гемолитические анемии).
2. Другие ХМПЗ и МПЗ/МДЗ.
3. С острыми лейкозами (в стадии бластного криза).
Критерием диагностики является наличие или отсутствие специфического для ХМЛ маркера Ph-хромосомы и bcr/abl.
Лечение. Несмотря на появление новых эффективных препаратов, таких как иматиниб, дазатиниб и нилотиниб, аллогенная трансплантация ГСК у детей и молодых больных (моложе 50 лет) по-прежнему является терапией выбора и позволяет излечить определенную группу больных.
Показатель выздоровления в группе больных с родственной аллогенной трансплантацией составляет 60%, с неродственной трансплантацией — около 50%.
При наличии у пациента потенциальных доноров необходимо решить вопрос о возможности аллогенной трансплантации, определяя уровень риска от трансплантации.
Наилучшие результаты показаны при ТГСК в хронической фазе, в первые 2 года после постановки диагноза.
Учитывая значительное снижение летальности при лечении гливеком (иматинибом), он может быть рекомендован всем больным в качестве первой линии терапии.
В хронической фазе доза гливека — 400 мг/день ежедневно, в фазе акселерации и бластного криза — 600—800 мг/день.
Гливек — ингибитор тирозинкиназы, механизм его действия заключается в блокировании активности белка р-210-bcr/abl-тирозинкиназы, играющей ключевую роль в патогенезе ХМЛ.
При назначении гливека в качестве первой линии терапии частота полных цитогенетических ответов через 12 мес лечения составляет 75—95%, в фазе акселерации — 24—17%, в фазе бластного криза - 16-7%.
Гидроксимочевина (гидреа, литалир) может назначаться в качестве первой линии терапии практически у всех больных для уменьшения массы опухоли на период обследования и решения вопроса о дальнейшей тактике лечения.
Доза гидреа определяется с учетом количества лейкоцитов и веса больного. При лейкоцитозе более 100х10*9/л — 50 мг/кг/день, в дальнейшем при снижении количества лейкоцитов дозу уменьшают: при лейкоцитозе (40-100)х10*9/л — 40 мг/кг/день, (20-40)х10*9/л - 30 мг/кг/день, (5-20)х10*9/л - 20 мг/кг/день.
Реаферон-а (Интрон А, Роферон А, Реаферон).
Применение реаферона позволяет увеличить сроки выживаемости по сравнению с химиотерапией (гидреа, бусульфан).
Оптимальная доза 5 млн/м2/день.
Для группы низкого риска 10-летняя выживаемость больных с полным цитогенетическим ответом составляет 100%, с большим цитогенетическим ответом — 76—78%, для остальных — 45—48%.
Бусульфан — в связи с появлением более эффективных для терапии ХМЛ препаратов, применение бусульфана в настоящее время ограничено. Необходимо отметить, что применение бусульфана в качестве первой линии терапии значительно ухудшает результаты трансплантации костного мозга.
Для терапии резистентных к гливеку пациентов в настоящее время используются и проходят стадию клинических исследований антитирозинкиназные препараты нового поколения дазатиниб и нилотиниб, по своей эффективности превышающие гливек.
Критерии гематологической ремиссии (оценивается по количеству лейкоцитов в периферической крови и выраженности спленомегалии): полная — лейкоциты 20х10*9/л, стойкая спленомегалия.
Критерии цитогенетического ответа (определяется по проценту выявляемых Ph-позитивных клеток в костном мозге): полный — Ph-позитивные клетки отсутствуют; большой — Ph—позитивные клетки 95%.
Прогноз.
Средняя продолжительность жизни пациентов в хронической фазе на фоне стандартной терапии составляет 5—7 лет и зависит от чувствительности к реаферону.
Трансплантация костного мозга позволяет излечить 50—60% больных, эффективность транслантации зависит от фазы заболевания.
Отдаленных результатов на терапии гливеком пока нет.
По статистике, заболеваемость ХМЛ составляет один-два случая на 100 000 населения в год. Основная группа пациентов – люди среднего возраста (чаще всего 30–50 лет), у детей же ХМЛ встречается редко, составляя не более 2–5 % от числа всех лейкозов.
Заболевание характеризуется повышенным образованием клеток крови лейкоцитов, особенно нейтрофилов, которые попадают во многие органы, в первую очередь селезенку. Без должной терапии хроническая стадия длится в среднем три – пять лет и в конечном итоге переходит в терминальные стадии (фаза акселерации и бластный криз), после чего довольно быстро наступает летальный исход.
История лечения больных с увеличенной селезенкой, необъяснимой слабостью, одышкой, потливостью, непереносимостью жары, бледной кожей, потерей веса насчитывает столетия. О первых успешных опытах терапии ХМЛ сообщили в Германии в 1865 году – немецкие врачи заявили об улучшении самочувствия и уменьшении объемов селезенки у двух пациентов, которым давали раствор мышьяка. Этот яд вплоть до начала ХХ столетия оставался единственным средством лечения хронического миелолейкоза. И все же увеличить продолжительность жизни больных с его помощью было невозможно – терапия мышьяком лишь на короткое время уменьшала проявления тяжелой болезни и облегчала состояние больных. Впоследствии таких пациентов пытались лечить с помощью бензола, уретана, радиоактивного фосфора. Эффект от этих средств был недолгим и незначительным, а переносились пациентами они плохо.
На заре прошлого века, в 1902 году, для лечения ХМЛ впервые с успехом применили облучение селезенки. Благодаря этой процедуре объемы органа сокращались, а неприятные симптомы исчезали. Казалось, эффективный способ наконец найден – вплоть до середины ХХ века эта терапия считалась единственным методом лечения ХМЛ. Но, увы, это давало лишь кратковременный эффект: в большинстве случаев пациенты теряли работоспособность уже на второй год после постановки диагноза, а еще через год становились лежачими инвалидами.
В течение десятилетий врачи считали болезнь неизлечимой, а терапия ХМЛ носила паллиативный (симптоматический) характер. Первой важной вехой на пути революции в лечении ХМЛ стало появление препарата на основе производной дисульфоновой кислоты алкирующего действия (торговые названия – бусульфан, милеран, миелосан). На многие годы этот препарат стал одним из вариантов выбора при лечении ХМЛ – он снимал симптомы заболевания, а пациенты на фоне лечения могли продолжать работать и вести привычный образ жизни. И все же поводов для полного оптимизма не было: через четыре – шесть лет лечения дело заканчивалось бластным кризом, и примерно через полгода пациент умирал.
Добиться длительных ремиссий у пациентов с ХМЛ ученым удалось в 80-е годы прошлого века, когда для терапии стал использоваться а-интерферон. Было доказано, что он эффективно подавляет Ph-позитивные клетки более чем у половины пациентов с ХМЛ, – это был новый прорыв в лечении заболевания. И все же часто переносимость этого препарата была плохой: 20 % пациентов вынуждены были прекращать лечение из-за серьезных побочных эффектов. Некоторые же больные на препарат не реагировали вообще. При этом с течением времени, пусть и длительного, болезнь неизбежно переходила в терминальную стадию.
Разработать принципиально новые подходы к лечению ХМЛ удалось благодаря развитию молекулярной биологии. Еще в 60-х годах прошлого века ученые Питер Ноуелл (Пенсильванский университет) и Дэвид Хангерфорд (Онкологический центр Фокса Чейза в Филадельфии) впервые расшифровали патогенез ХМЛ, обнаружив соматическую мутацию в стволовой кроветворной клетке, в результате которой происходит обмен генетическим материалом между хромосомами 9 и 22. Это приводит к образованию филадельфийской, или Ph-хромосомы с онкогеном BCR-ABL, который играет ключевую роль в нарушении жизнедеятельности клеток: из нормальных они превращаются в злокачественные. Сегодня филадельфийскую хромосому можно обнаружить при помощи стандартного цитогенетического исследования, или метода FISH.
В результате мутации в организме вместо двух белков синтезируется один, который продуцируется измененной хромосомой, – тот самый аномальный белок BCR-ABL. Он функционирует как фермент тирозинкиназа, из-за которого клетки начинают безудержно расти и делиться, что приводит к образованию огромного количества лейкоцитов в организме, способных влиять на функционирование различных органов.
Киназы правят жизнью клетки, вовремя запуская нужные процессы. Но если ход их работы нарушается, последствия печальны. Чрезмерная активность киназ лежит в основе многих онкологических заболеваний. Кстати, первую тирозинкиназу ученые открыли в 1980 году, изучая онкологическое заболевание кур – саркому Рауса.
О том, что пусковым механизмом развития ХМЛ является активация ABL-тирозинкиназы, стало известно в 1990-е годы. Тогда ученые принялись разрабатывать средство, позволяющее подавить патологическую активность онкопротеина BCR-ABL, то есть ингибитор тироксинкиназы.
Применение нового лекарства было разрешено в США в мае 2001 года после двухлетних клинических испытаний. Создание препарата строго направленного патогенетического действия стало большим прорывом в борьбе с лейкозом, подобного которому раньше не было. Его появление кардинально изменило подходы к лечению ХМЛ и прогноз таких больных. Терапия достоверно приводит к пятилетней выживаемости у 90 % пациентов против 30–40 % при лечении цитостатическими препаратами.
Данный текст является ознакомительным фрагментом.
- Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Scince 1960;132:1497.
- Rowley JD. А new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa banding. Nature 1973;243:290.
- Selected Proceedings of the First European School of Haematology Course of Chronic Myelogenous Leukemia. Biother Update 1997;1:1.
- Van Etten RA. Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol 1999;9:179.
- Mayer BJ, Baltimore D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol Cell Biol 1994;14:2883.
- Verfaillie CM. Biology of chronic myelogenous leukemia. Hematology/Oncology Clin North Am 1998;12:1-29.
- Zhao RC, Tarone G, Verfaillie CM. Presence of the adhesion inhibitory b1B integrin isoform on CML but not normal progenitors is et least in part responsible for the decreased CML progenitor adhesion. Blood 1997;90:393a.
- Reiter A, Sohal J, Kulkarni S, et al. Consistent fusion of ZNF198 to the fibroblast growth factor receptor-1 in the t(8;13)(p11;q12) myeloproliferative syndrome. Blood 1998;92:1735.
- Kennedy BJ, Yarbro KW. Metabolic and therapeutic effects of hydroxyurea in chronic myeloid leukemia. JAMA 1966;195:1038.
- Kennedy BJ. Hydroxyurea in chronic myelogenous leukemia. Ann Intern Med 1969;70:1084.
- Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. Blood 1993;82:398.
- Talpaz M, Kantarjian H, McCredie K, et al. Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha A in chronic myelogenous leukemia. N Engl J Med 1986;314:1065.
- Allan N, Richards S, Shepherd P, et al. Medical Research Council randomized multicenter trial of intrferon a n1 for chronic myeloid leukemia. Lancet 1995;345:1392.
- Bhatia R, Munthe H. Verfaillie CM. Decreased b 1-integrin receptor capping in CML progenitors reflecting abnormal receptor cytoskeletal interactions is normalized by interferon-a. Blood 1996;88:2537a.
- Cortes JE, Talpaz M, Kantarjian H. Chronic myelogenous leukemia: a review. Am J Med 1996;100:555-70.
- Sokal JE, Leong SS, Gomez GA. Preferential inhibition by cytarabine of CFU-GM from patients with chronic granulocytic leukemia. Cancer 1987;99:197.
- Guilhot F, Chastang C, Michallet M, et al. Interferon - alfa 2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. N Engl J Med 1997;337:223.
- Goldman JM. Stem Cell Transplantation for CML: Its Place in Treatment Algorithms – 2001. In: Chronic Myelogenous Leukemia. Haematology 2001;103.
- Druker BJ, Tamura S, Buchdunger E, et al. Effects of selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561.
- Schinder T, et al. Structural mechanism for STI571 inhibition of Abelson tyrosine kinase. Science 2000;289:1938.
- Glivec (imatinib) Summery of Product Characteristics. Basel, Switzerland: Novartis Pharma AG 2001.
- Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031.
- Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001;344:1038.
- Glivec. Clinical Monograph 2001, Novartis Pharma.
- Druker BJ, for the IRIS Study Group. STI571 (Gleevec/Glivec, imatinib) versus interferon (IFN) + cytarabine as initial therapy for patients with CML: results of a randomized study. Prog/Proc ASCO 2002;21(part 1, p 1a):No 1.
- Hahn EA. The quality of life of patients with chronic phase chronic myeloid leukemia in the IRIS study of interferon-alpha plus ara-c vs imatinib (STI571, Glivec). Hematology J 2002;3(suppl. 1):300.
- Kantarjian H, Jorge C, O’Brien S, et al. High rates of early major and complete cytogenetic responce with imatinib mesylate therapy given at 400 mg or 800 mg orally daily in patients with newly diagnosed philadelphia chromosome-positive chronic myeloid leukemia in chronic phase. Prog/Proc ASCO 2002;21(part 1, p 261a):No 1043.
- Cortes JE, Talpaz M, Giles FJ, et al. High-dose imatinib mesylate (STI571, Gleevec) in patients with chronic myeloid leukemia (CML) resistant or intolerant to interferon-alpha (IFN). Prog/Proc ASCO 2002;21(part 1, p 262a):No 1044.
- Hochhaus A., Lahaye T., Kreil S, et al. Interim analysis of imatinib treatment in 300 patients with chronic myelogenous leukemia (CML): evaluation of response and resistance. Prog/Proc ASCO 2002;21(part 1, p 262a):No 1045.
- Gately K, McCann SR, Caroll P, et al. Cytogenetic and molecular analysis of BCR- ABL expression in CML patients following STI571 (Glivec) treatment. he Hematology Journal 2002;3(suppl. 1):296.
- Martinelli G, Glannini B, Rosti G, et al. CML patients with complete karyotypic response to STI571 (Glivec) therapy show rapid redaction of BCR-ABL transcripts. On behalf of the Italian cooperative study group on CML. Hematology J 2002;3(suppl. 1):297.
- Kontsioti F, Pappa V, Papasteriadis C, et al. In vitro effect of STI571 in combination with A-interferon on the proliferation, differentiation and apoptosis of K562 cells. Hematology J 2002;3(suppl. 1):290.
- Korycka A, Robak T. The comparison of influence of STI571 used alone or in combination either with 2-chlorodeoxyadenosine or fludarabine on the normal and chronic myelogenous leukemia progenitor cells in vitro. Hematology J 2002;3(suppl. 1):292.
- O’Dwyer ME, Mauro MJ, Aust S, et al. Ongoing evaluation of the combination of imatinib mesylate (Glivec) with low dose interferon-alpha for the treatment of chronic phase of CML. Hematology J 2002;3(suppl. 1):293.
- Grasso R, Miglino M, Varaldo R, et al. Restoration of polyclonal hematopoiesis in STI 571-induced complete cytogenetic remission of chronic myelogenous leukemia. Hematology J 2002;3(suppl. 1):299.
- Biernaux C, Loos M, Sels A, et al. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995;86:3118.
- Bose S, Deininger M, Gora-tybor J, et al. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood 1998;92:3362.
- Gratwohl A, Hermans J, Goldman JM, et al. Risk assessment for patients with chronic myeloid leukemia before allogenic blood or marrow transplantation. Lancet 1998;352:1087.
- Martinez J, de la Rubia J, Sanz G, et al. Unrelated cord blood transplantation in adult patients with chronic mieloid leukemia on blastic phase after responce to STI 571. Hematology J 2002;3(suppl. 1):291.
- Carella AM, Beltrani G, Rossi E, et al. Safety and efficacy of STI 571 in patients with CML relapsed after autograftig (ASCT) and IFN-A. Hematology J 2002;3(suppl. 1):295.
- Beham-Schmid Ch, Apfelbeck U, Sill H, et al. Treatment of chronic myelogenous leukemia with the tyrosine kinase inhibitor STI571 results in marked regression of bone marrow fibrosis. Blood 2002;99:381.
Читайте также: