
Т клеточный пролимфоцитарный лейкоз прогноз
Хронический лимфоцитарный лейкоз (ХЛЛ) является одной из разновидностей рака крови. Его также называют хронической лимфоидной лейкемией или лимфомой малых лимфоцитов.
Что такое хронический лимфоцитарный лейкоз?
ХЛЛ развивается из-за аномалий в образовании и развитии одной из разновидностей кровяных телец – лимфоцитов.
Большинство случаев ХЛЛ (около 95%) начинается с повреждения B-лимфоцитов (B-клеток). Основные признаки:
Из-за этого лейкоциты не могут нормально выполнять некоторые свои функции по борьбе с инфекциями;
Постепенно они накапливаются в костном мозге и крови, вытесняя из кровотока здоровые лимфоциты;
Низкий уровень здоровых лимфоцитов может привести к заражению вторичными инфекциями, анемии и кровотечениям;
Поврежденные клетки разносятся кровотоком по всему телу и мешают нормальному функционированию органов;
В редких случаях хроническая форма лейкоза переходит в агрессивную.
Другие разновидности хронических лимфом
Помимо ХЛЛ существуют и другие разновидности лейкозов.
Пролимфоцитарная лейкома (ПЛЛ). Она более агрессивна, чем большинство типов ХЛЛ. Поражает как B-лимфоциты, так и T-лимфоциты. Обычно развивается стремительнее ХЛЛ, но все-таки не так быстро, как острый лимфобластный лейкоз.
Крупнозернистая лимфоцитарная лейкемия (КЛЛ). Имеет тенденцию к медленному росту, однако, в некоторых случаях быстро переходит в агрессивную стадию. Характеризуется увеличенными лимфоцитами с видимыми гранулами, поражает T-лимфоциты или естественные киллеры (NK-клетки).
Волосатоклеточный лейкоз (ВКЛ). Медленно растущая разновидность рака B-клеток, при этом довольно редкая. Название происходит от внешнего вида лимфоцитов – точечных проекций на поверхности клеток, которые делают их волосатыми на вид.
Малая лимфоцитарная лимфома (МЛЛ). Это заболевание тесно связано с хронической формой лимфомы, однако, при МЛЛ раковые клетки обнаруживаются в лимфоузлах и селезенке, а не в костном мозге и крови.
Органы кроветворения и ХЛЛ
Для начала полезно разобраться, как вообще устроено кроветворение в организме и какую роль в этом играет костный мозг.
Стволовыми клетками называют особый тип клеток в организме, которые могут трансформироваться практически в любую иную форму: клетки печени, кожи, мозга или крови. Они формируются в костном мозге. Те стволовые клетки, которые участвуют в кроветворении, называются гемопоэтическими (стволовыми клетками крови).
Кровяные тельца непрерывно стареют, повреждаются и погибают. На них место должны непрерывно поступать новые, причем в достаточном количестве. Так, например, в норме у здорового взрослого человека должно содержаться от 500 до 1500 лимфоцитов на 1 мкл (примерно 25-40% от общего объема крови).
Стволовые клетки продуцируются в основном в мягкой губчатой ткани костей, но некоторое их число также можно встретить в циркулирующей крови.
Гемопоэтические клетки активно трансформируются в лимфоидные и миелоидные стволовые клетки:
-
Лимфоидные вырабатывают лимфобласты, которые в свою очередь преобразуются в несколько типов лейкоцитов, включая лимфоциты и NK-клетки;
Миелоидные, соответственно, продуцируют миелобласты. А те превращаются в другие типы лейкоцитов: гранулоциты, эритроциты и тромбоциты.
У каждого типа клеток крови своя специализация и предназначение.
Лейкоциты активно противостоят инфекциям и внешним раздражителям.
Красные кровяные тельца (эритроциты) отвечают за перенос кислорода из легких к тканям и доставку углекислого газа обратно в легкие для удаления.
Тромбоциты образуют сгустки, чтобы замедлить или прекратить кровотечение.
На ранних стадиях ХЛЛ обычно не беспокоит пациента. Для развития выраженных симптомов могут уйти годы, но как только они появляются – это уже повод говорить о хронической стадии заболевания.
Симптомы ХЛЛ часто путают с гриппом и прочими распространенными заболеваниями. При этом падает уровень содержания всех типов клеток крови. Симптомы низких лейкоцитов в крови:
жар, потливость, боли в различных частях тела;
Также может наблюдаться снижение уровня эритроцитов:
усталость, слабость, недостаток энергии и сонливость.
Симптомы низких тромбоцитов:
красные пятна нёбе или лодыжках;
частое или сильное носовое кровотечение;
синяки по всему телу и плохая свертываемость крови при порезах.
Общие симптомы хронического лимфоцитарного лейкоза:
необъяснимая потеря веса;
боли в костях или суставах;
опухание лимфатических узлов в шее, подмышках, желудке или паху.
Диагностика хронического лимфоцитарного лейкоза
Вышеперечисленные симптомы уже могут навести вашего врача на подозрения, однако, чтобы поставить окончательный диагноз, ему потребуется изучить историю болезни и провести полное медицинское обследование.
Для точной диагностики ХЛЛ потребуется несколько тестов. Некоторые из них могут не понадобиться, но будут нужны, чтобы уточнить диагноз и разработать более эффективную стратегию лечения.
Тест на типы и количество клеток крови, наличие аномальных лимфоцитов или уже сформировавшихся раковых клеток. Врачу здесь необходимо определить тип дефектных клеток, признаки замедления или наоборот прогрессирования рака. Применяется два вида специальных анализов крови: имунофенотипирование и проточная цитометрия. Иногда ХЛЛ можно заподозрить и при помощи общего анализа.
Отбор тканей из костей таза при помощи иглы (аспирация и биопсия костного мозга) и их проверка на наличие раковых клеток.
Клетки крови или костного мозга проверяют на наличие хромосомных аномалий: недостающих частей, дополнительных копий, дублировании хромосом. Также могут быть проверены изменения в белках иммунной системы, которые могут предсказать степень агрессивности ХЛЛ. В целом, выделяют три вида генетических тестов: цитогенетический анализ, флуоресцентную гибридизацию in situ (FISH)и полимеразную цепную реакцию (ПЦР-тест).
Сюда входят рентген грудной клетки, компьютерная и магнитно-резонансная томографии, а также ультразвуковое исследование лимфоузлов.
Стадии хронического лимфоцитарного лейкоза по Rai
Чтобы определить, как далеко зашла болезнь, и спланировать лечение, врачи пользуются системой Rai. Она была специально разработана для ХЛЛ:
Стадии заболевания зависят от количества лимфоцитов, эритроцитов и тромбоцитов в костном мозге и кровотоке, а также от того, были ли поражены селезенка, печень и лимфоузлы;
Стадии варьируются от 0 до IV, где 0 – наименее, а IV – наиболее тяжелая.
Ваш этап Rai даст онкологу информацию о вероятности прогрессирования болезни и необходимости лечения. Этап 0 характеризуется низким уровнем риска, этапы I–II – умеренным, этапы III–IV – высоким.
Ваш врач должен тщательно изучить и другие факторы, чтобы спрогнозировать перспективы и подобрать наиболее оптимальную стратегию лечения. В их числе:
Генетические отклонения и мутации в лейкоцитах (к примеру, отсутствие части хромосомы или наличие дополнительной хромосомы);
Наличие мутационного статуса IGHV-гена (наличие тяжелых цепей иммуноглобулина с переменной областью);
Проявляются ли симптомы ХЛЛ;
Возраст, наличие сопутствующих заболеваний, образ жизни;
Количество онкогенных (прелейкемических) клеток;
Скорость деления лейкемических клеток;
Как болезнь реагирует на первичное лечение и как долго длится ответная реакция.
Онкологу потребуется провести дополнительные тесты, анализы крови и костного мозга после анализа лечения.
Ремиссия будет означать, что болезнь реагирует на терапию. Полная ремиссия означает отсутствие каких-либо симптомов и клинических признаков рака. Частичная ремиссия означает уменьшение всех симптомов на 50%;
Рецидив означает, что ХЛЛ возвращается после пребывания в ремиссии более шести месяцев;
Резистентность – заболевание прогрессирует в течение шести месяцев после лечения.
Насколько распространен хронический лимфоцитарный лейкоз?
В США ежегодно диагностируется порядка 20 тысяч случаев ХЛЛ. По статистике это наиболее распространенная разновидность лейкемии среди взрослых – на нее приходится почти 40% случаев.
Причины хронического лейкоза
Медицине не известно, что является причиной ХЛЛ. Известно, что болезни подвержены люди среднего и старшего возраста. Средний возраст пациентов на момент постановки диагноза составляет 72 года. ХЛЛ чаще встречается среди мужчин, чем среди женщин.
В целом, это заболевание более распространено в Северной Америке и Европе, чем в Азии. Однако это зависит не от места проживания, а скорее от генетической предрасположенности отдельных рас. Азиаты, живущие в США, Канаде или европейских странах, подвергаются примерно одинаковому риску ХЛЛ со своими соплеменниками из азиатских стран.
В настоящий момент выявлено всего два фактора риска по ХЛЛ:
случаи ХЛЛ или иных типов лейкозов среди близких родственников.
Нужно иметь в виду, что у многих людей с ХЛЛ вообще не было факторов риска в анамнезе.
В середине 70-х годов XX в. одновременно с открытием иммунологических маркеров В- и Т-лимфоцитов появилась возможность разделения этих клеточных популяций. Соответственно двум типам лимфоцитов были выделены две основные группы лимфопролиферативных заболеваний: В- и Т-клеточные. Первый иммунологический тест, позволивший продемонстрировать Т-клеточную природу лимфоцитов, основывался на способности этих клеток формировать розетки при инкубации с эритроцитами барана (Е-розетки).
Совершенствование лабораторных методов исследования, особенно иммунологических и молекулярно-генетических, позволило более детально охарактеризовать и классифицировать эту группу заболеваний. Т-клеточные лимфопролиферативные заболевания составляют 10—15 % от всех опухолей лимфатической системы и по уровню дифференцировки и созревания могут быть разделены на две группы: тимические и посттимические.
Посттимические опухоли представлены иммунологически зрелыми Т-лимфоцитами, в ядрах которых отсутствует фермент терминальная дезоксинуклеотидилтрансфераза (TdT).
Одной из форм зрелоклеточных Т-клеточных лимфопролиферативных заболеваний является Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ), впервые описанный D. Catovsky и соавт. в 1973 г.. Наиболее часто Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) встречается в двух возрастных группах: пожилых (средний возраст 69 лет) и молодых с атаксией-телеангиэктазией (AT). Женщины болеют чаще, чем мужчины, в соотношении 4:1. Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) характеризуется агрессивным течением с медианой выживаемости 7,5 мес.
Заболевание начинается остро или подостро. Больные жалуются на быструю утомляемость, слабость, потливость, снижение массы тела. Одними из первых симптомов могут быть боли в животе, связанные с выраженной спленомегалией, увеличением внутри-брюшных лимфатических узлов, а также гематологические изменения (анемия, тромбоцитопения), обусловленные костно-мозговой недостаточностью и гиперспленизмом. Реже первой манифестацией заболевания является поражение кожи, отличающееся полиморфной картиной, — от кожной сыпи, обычно пятнисто-папулезной, до генерализованной эритродермии.
Органные поражения, например ЦНС и легких, встречаются редко. Менее чем у 5 % больных заболевание начинается бессимптомно, и только в анализе крови обнаруживается медленно нарастающий абсолютный лимфоцитоз. Такие случаи, особенно мелкоклеточный вариант Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ), ошибочно диагностируют как хронический лимфолейкоз (ХЛЛ). В противоположность ХЛЛ, течение которого может оставаться стабильным длительное время, Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) прогрессирует в течение нескольких месяцев.
Характерным лабораторным изменением при Т-ПЛЛ является высокий лейкоцитоз, который может достигать 1000 • 10 9 /л.
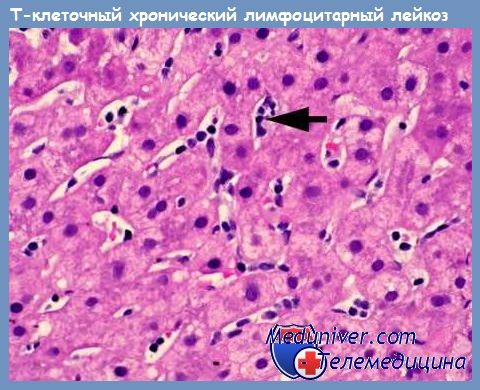
По данным обследования более 100 пациентов Е. Matures и соавт. представили основные клинико-лабораторные проявления Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ): спленомегалия — 73 %; лимфаденопатия — 53 %; гепатомегалия — 40%; поражение кожи — 27 %; лейкоцитоз более 100 • 109/л — 75 %; анемия и тромбоцитопения — 30 %.
Этиологические причины Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ) не установлены. В сыворотке крови больных не обнаружены антитела к вирусам HTLV-I/II даже у пациентов из эндемичных регионов. С помощью анализа ДНК не удалось доказать наличие геномной последовательности вируса HTLV-I в опухолевых клетках.
Морфологическим субстратом Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ) более чем в 2/3 случаев являются пролимфоциты. Они имеют несколько больший по сравнению с обычным лимфоцитом размер, ядро с конденсированным хроматином, которое часто имеет неровные очертания и нуклеолу. Цитоплазма опухолевых клеток базофильная и не содержит гранул. У 20 % больных Т-ПЛЛ опухолевые клетки меньшего размера, в большинстве из них при световой микроскопии нуклеола видна плохо, однако электронно-микроскопическое исследование позволяет выявить ультраструктурные особенности, присущие пролимфоцитам. Такие случаи относят к мелкоклеточному варианту Т-ПЛЛ.
При иммунологическом фенотипировании пролимфоциты, как правило, имеют иммунофенотип CD2+CD5+ и выраженно экспрессируют антиген CD7. Количество CD7-антигенных детерминант на поверхности опухолевых клеток при Т-клеточном пролимфоцитарном лейкозе (Т-ПЛЛ) значительно больше, чем на нормальных Т-лимфоцитах и лимфоцитах при других посттимических Т-клеточных лимфопролиферативных заболеваниях. В 20 % случаев на мембране пролимфоцитов не экспрессируется CD3, однако этот маркер всегда обнаруживается в цитоплазме клеток. Применительно к экспрессии CD4 и CD8 уникального фенотипа, присущего исключительно Т-ПЛЛ, не существует.
Чаще всего (2/3 случаев) пролимфоциты CD4+CD8-, редко фенотип клеток CD8+CD4- и приблизительно в 25 % определяется коэкспрессия CD4 и CD8.
Цитогенетические исследования, проведенные при Т-клеточном пролимфоцитарном лейкозе (Т-ПЛЛ), позволили выявить аномалии хромосом 14, 8 и 11. Перестройки хромосомы 14 составляют 2/3 всех цитогенетических изменений: инверсия хромосомы 14 — invl4(q11q32), тандемная транслокация между двумя хромосомами t(14; 14). Инверсия хромосомы 14 крайне редко встречается при других зрелоклеточных лимфопролиферативных заболеваниях Т-клеточной природы и считается патогномоничной для Т-клеточного пролимфоцитарного лейкоза. Важно отметить сходство цитогенетических изменений в опухолевых клетках при Т-ПЛЛ и в Т-лимфоцитах больных атаксией-телеангиэктазией. Описанные случаи развития Т-клеточных лейкозов у этих пациентов относятся к Т-ПЛЛ.
Как отмечалось, Т-клеточный пролимфоцитарный лейкоз является заболеванием с агрессивным течением. Больные Т-ПЛЛ обычно резистентны к стандартным схемам лечения, включающим алкилирующие препараты (хлорамбуцил, циклофосфамид). Включение в схему терапии антрациклинов (CHOP) позволяет получить ответ, чаще всего частичный и непродолжительный, только у 1/3 больных. Одним из наиболее активных цитостатических препаратов в лечении Т-клеточного пролимфоцитарного лейкоза является 2-деоксикоформицин (пентостатин). Использование его в дозе 4 мг/м2 еженедельно до достижения максимального эффекта позволяет получить общий ответ в 40 % случаев и только в 12 % случаев достигается полная ремиссия. В последние годы предпринимаются успешные попытки использовать при Т-ПЛЛ анти-CD52 моноклональное антитело (Campath-1H).
Иммунотерапия Campath-1H позволяет получить полную ремиссию более чем у половины пациентов, включая резистентных к деоксикоформицину.

-
5 минут на чтение

- Что это такое
- Причины
- Неблагоприятный радиационный фон
- Химические соединения
- Вирусные возбудители
- Генетические патологии
- Симптомы
- Стойкое малокровие
- Геморрагический синдром
- Гиперпластические нарушение внутренних органов
- Болевой синдром
- Появление лейкемид
- Диагностирование
- Лечение
- Осложнения
- Выживаемость
- Профилактика Т-клеточного лейкоза
Что это такое
Т-клеточный лейкоз известен под названием лимфома. Он представляет собой раковую опухоль, состоящую из лимфоцитов категории CD-4. Чаще всего она развивается по причине патогенного воздействия вируса Т-клеточного лейкоза.
При отсутствии надлежащего лечения прогрессирование болезни ведёт к нарушениям работы органов, систем и кожных покровов и разрушению суставов и костей. Возможно появление гиперкальциемии. Во время диагностирования в крови пациента обнаруживаются атипично изменённые лимфоциты.
Наиболее высокий порог распространённости отмечается в южной части Японии, на территории островов Карибского бассейна, на берегах Тихого океана и среди жителей Африканского материка. Мужская половина человечества подвержена к развитию данного типа лейкоза больше, чем женщины. Особенно подвержены заражению наркоманы и иные лица, ведущие асоциальный образ жизни.
Причины
Т-клеточный лейкоз развивается при наличии сразу нескольких факторов.
Роль радиационного излучения в провоцировании возникновения рака крови подтверждается на основании выявления внезапного скачка заболеваемости лейкозом среди жителей Японии, после ядерного поражения городов Хиросима и Нагасаки.
Также имеются сведения о повышении числа больных с Т-клеточным лейкозом среди людей, получавших ранее курс радиотерапии в целях борьбы с имеющимися у них онкологическими новообразованиями.
Наиболее ярко выраженным онкогенным воздействием обладают нефтепродукты. Эти вещества широко используются в индустриальных целях. Соединения бензола и иных нефтепродуктов легко попадают внутрь человеческого тела во время дыхания и сквозь кожные покровы.
Это химическое вещество скапливается в нервных и жировых тканях. Другими химическими соединениями, влияние которых способно приводить к формированию лейкемии, являются пестициды, содержащие галогены, моющие средства и лакокрасочная продукция.
Также установлено, что причиной развития лейкемии могут стать химиотерапевтические фармацевтические вещества, применяющиеся в медицине в целях борьбы с лимфогранулематозом.
Вирус Т-клеточного лейкоза имеет в себе гены, способные при соприкосновении со здоровыми лейкоцитами крови превращать их в раковые.
Лейкозы тесно связаны с наличием таких генетических патологий, как врожденные расстройства деятельности иммунной системы. Поэтому немалую значимость в развитии Т-клеточного лейкоза играет наследственный фактор.
Симптомы
У подавляющего большинства лиц с лейкозом на начальной стадии развития комбинация симптомов может отличаться. Количество и интенсивность симптомов в каждом конкретном случае будет зависеть от состояния иммунной системы и уровня общей физической подготовки человека.
И всё же, в большинстве случаев люди, страдающие Т-клеточным лейкозом, отмечают у себя один или несколько из симптомов.
Это состояние вызывается подавлением процесса выработки эритроцитов. Она сочетается со стойким незначительным повышением температуры тела, постоянной подавленностью и быстрой утомляемостью, регулярными приступами общего недомогания, потерей сознания и предобморочными состояниями.
Малое количество зрелых тромбоцитов становится причиной кровоизлияний на кожных покровах и слизистых оболочках, которые могут принимать форму как мелких вкраплений, так и обширных участков. Запущенный геморрагический синдром может стать причиной необъяснимых синяков на коже и сильных кровотечений.
Атипично изменённые лейкоциты приводят к увеличению размеров лимфатических узлов, а также печени и селезёнки.
Продолжительная интоксикация организма становится причиной постоянных болей в суставах и костях. Из-за расстройства функционирования иммунной системы больные лейкемией чаще обычного страдают вирусными, инфекционными и бактериальными заболеваниями, что также усугубляет проявления болевого синдрома.
Признаком запущенной стадии лейкоза принято считать появление на поверхности кожных покровов лейкемид – твердых коричневых либо красноватых пятен.
Диагностирование
При начальной стадии течения болезни зачастую раковых клеток в крови ещё очень мало. Чтобы выявить болезнь на ранней стадии, необходимо расширенное исследование клеток гранулярных лейкоцитов при помощи специальных микроскопов.
Если результат исследования выявит, что количество гранулярных клеток существенно превышает установленную норму, то для подтверждения диагноза больному будет назначена проточная цитометрия крови.

- Онкогематология
![]()
Наталья Геннадьевна Буцык- 6 декабря 2019 г.

Процент схожести лейкоцитов определяют с помощью исследования Т-клеточного рецептора – молекуле, отличающей отдельный лейкоцит от всей остальной лейкоцитарной массы. Проводят данное исследование в лабораториях молекулярно-генетических исследований.
С целью подтверждения наличия заболевания пациенту могут быть назначены дополнительные исследования. В их числе общий анализ крови, патоморфологичское исследование кожных покровов, биохимический анализ, иммуно-ферментный анализ крови и иммунный блоттинг.
Лечение
В наше время пользуется популярностью такой способ лечения Т-клеточного лейкоза, как химиотерапевтическая терапия, заключающаяся в приёме сильнодействующих фармацевтических препаратов. Эти лекарства должны приниматься курсом в виде таблеток и капсул, назначенных лечащим врачом. При отсутствии результатов лечение пациента будет проводиться в стационаре с помощью внутривенных либо внутримышечных инъекций
Ещё один действенный метод – это лучевая терапия. Данный способ лечения лейкозов включает в себя применение рентгеновских лучей или иных видов радиационного излучения.
При необходимости пациенту может быть проведено трансплантирование стволовых клеток. Данный вид вмешательства предназначается для замены патологически изменённых лейкоцитов крови здоровыми клетками.
Осложнения
Анемия – неизменное последствие пациентов, страдающих лейкозом. Данное заболевание формируется потому, что поражение костного мозга постепенно сводит на нет возможности для нормального процесса кроветворения.
Эритроциты формируются из лейкемических нормобластов, лейкоз же нарушает процесс нормального формирования эритроидных клеток – предшественников эритроцитов. Всё это приводит к малоэффективному протеканию гемопоэза, из-за чего эритроциты циркулируют в крови более короткий срок.
Нередко у пациентов, страдающих Т-клеточным лейкозом, отмечается и тромбоцитопения. Причина данного состояния связана с поражением костного мозга и резким снижением активности тромбоцитов, что постепенно приводит к появлению геморрагических проявлений.

- Онкогематология
![]()
Ольга Владимировна Хазова- 5 декабря 2019 г.

Через определённый период времени из-за неполноценности лейкоцитов у пациентов развивается существенное снижение иммунитета, что приводит к повышению чувствительности перед вирусными, микозными и бактериальными возбудителями.
Геморрагические проявления приводят к увеличению печени и селезёнки. При запущенных стадиях заболевания нередко отмечается инфаркт селезенки, требующей отдельной симптоматической терапии.
В числе наиболее частых осложнений при средних и тяжёлых стадиях лейкозов возможно также появление инфильтраций во внутренних органах и в спинном мозге, что приводит к серьёзным нарушениям функционирования различных органов и систем.
В наиболее запущенных стадиях Т-клеточного лейкоза возможно формирование такого состояния, как гиперпуринемия.
Последующая стадия лейкоза — нефропатия, в наиболее запущенной стадии у пациентов нередко отмечается бластный криз.
Выживаемость
Многие дети до 5 лет с диагнозом Т-клеточный лейкоз при наличии должного лечения живут весь период борьбы с болезнью без каких бы то ни было осложнений. В случае если по завершении лечения заболевание никак не проявляет себя в течении 5 лет и более, ребёнок может считаться выздоровевшим.
При проявлениях рецидивирующего Т-клеточного лейкоза достичь ремиссии может быть несколько сложнее. При повторном возникновении болезни спасти жизнь ребёнку и стабилизировать его состояние здоровья в силах трансплантации спинного мозга, которая помогает вторично победить рак у 65%.
Опыт показывает, что одновременное проведение Т–клеточной иммунотерапии и лучевого лечения не только продлевает жизнь людям, но и в разы уменьшает скорость прогрессирования онкологического заболевания и возможных осложнений.
Кроме того, успех лечения зависит от целого ряда факторов, включая эмоциональное состояние самого больного, а также его близких родственников и друзей. При наличии поддержки и умении сохранять положительный настрой шансов на излечение всегда больше.
Профилактика Т-клеточного лейкоза
Обратиться за врачебной помощью следует в случае выявления одного или нескольких симптомов лейкоза. Для недопущения дальнейшего распространения заболевания крови стоит обследовать всех членов семьи, а также половых партнеров больного. Следует помнить, что лица, страдающие Т-клеточным лейкозом, либо страдавшие им в прошлом, не должны быть в числе доноров.
Что такое Пролимфоцитарный лейкоз -
Пролимфоцитарный лейкоз - вариант хронического лимфоцитарного лейкоза с более крупными и менее дифференцированными клетками.
Что провоцирует / Причины Пролимфоцитарного лейкоза:
Патогенез (что происходит?) во время Пролимфоцитарного лейкоза:
Различают следющие варианты пролимфоцитарного лейкоза:
- В-клеточный вариант - наиболее распространен.
- Т-клеточный вариант - встречается редко; субпопуляции хелперов обнаруживаются, возможно, чаще, чем супрессорные субпопуляции.
Симптомы Пролимфоцитарного лейкоза:
Диагностика Пролимфоцитарного лейкоза:
При пролимфоцитарном лейкозе в периферической крови и костномозговом пунктате преобладают (более 55%) пролимфоциты. Патологические клетки у 75-80% больных имеют В-клеточный фенотип, которые по своим биологическим характеристикам являются более зрелыми лимфоидными элементами, чем лимфоциты при типичном В-клеточном хроническом лимфолейкозе. У 20-25% больных клетки имеют Т-клеточный фенотип, в таких ких случаях заболевание протекает более тяжело, с выраженным лейкоцитозом, быстро прогрессирует, терапия мало эффективна.
Характерная особенность пролимфоцитарного лейкоза - выраженные лейкоцитоз и спленомегалия при незначительно увеличенных лимфоузлах, а также устойчивость к цитостатикам.
Лечение Пролимфоцитарного лейкоза:
Пролимфоцитарный лейкоз, как правило, резистентен к полихимиотерапии.
Профилактика Пролимфоцитарного лейкоза:
К каким докторам следует обращаться если у Вас Пролимфоцитарный лейкоз:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Пролимфоцитарного лейкоза, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Euro lab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Euro lab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Иммунофенотипирование.
Опухолевые клетки экспрессируют мембранные В-клеточные антигены (CD19, CD 20, CD22, CD79a, CD25, CD103, CD11c, HLA-DR), поверхностные и цитоплазматические иммуноглобулины.
Цитогенетика.Наиболее часто встречается трисомия 5 хромосомы.
Пролимфоцитарный лейкоз описан в 1974 г. Galton. Наиболее часто встречается В-клеточный фенотип опухолевых клеток (80% наблюдений), реже – Т-клеточный. Заболевание в основном регистрируется у пожилых мужчин
В-пролимфоцитарный лейкоз
В костном мозге наблюдается диффузная лимфоидная инфильтрация
В гемограмме имеет место гиперлейкоцитоз (более 100Х109/л), анемия, тромбоцитопения. В лейкоцитарной формуле пролимфоциты составляют более 55%
Морфология пролимфоцитов – крупные клетки диаметром 10-15 мкм. с округлым ядром, конденсированным хроматином, одной крупной нуклеолой
цитоплазмой базофильного цвета
Иммунологический фенотип.В-пролимфоциты характеризуются экспрессией В-клеточных антигенов – CD19, CD20, CD24. В отличие от В-ХЛЛ клетки имеют высокую плотность поверхностных иммуноглобулинов, значительно реже экспрессируют CD5
ЦитогенетикаПриблизительно 60% больных имеют патологию в области 14 хромосомы – 14q+. Реже встречается трисомия 12 хромосомы и транслокация t(6;12)(q15;p13).
Клиническими особенностями В-пролимфоцитарного лейкоза являются:
· •Незначительное увеличение лимфатических узлов.
Средняя продолжительность жизни около 3 лет
Т-пролимфоцитарный лейкоз
•В костном мозге диффузная лимфоидная инфильтрация с преобладанием пролимфоцитов.
В периферической крови анемия, гиперлейкоцитоз с пролимфоцитами, по морфологии не отличающимися от таковых при В-пролимфоцитарном лейкозе
Иммунологический фенотипКлетки чаще соответствуют Т-хелперам.
•Антигены: CD2+, CD3+, CD5+, CD7+, CD4+, CD8-/+. Характерна положительная цитохимическая реакция на a-нафтилацетатэстеразу в виде больших гранул
Клинические особенности•Заболевание протекает агрессивно, трудно поддается лечению. Часто значительно выражено поражение кожи (эритемы, папулы и др).
Грибовидный микоз (ГМ) относится к первичным Т-клеточным лимфомам кожи. Заболевание встречается у лиц старше 50 лет. Патогенез заболевания связан со злокачественной пролиферацией Т-лимфоцитов в коже с последующим поражением висцеральных органов, лимфатических узлов и костного мозга
Клиника.Начальные проявления ГМ не сопровождаются поражением лимфатических узлов и костного мозга.
Классическая форма ГМ имеет 3 стадии: эритематозную, бляшечную и опухолевую. Деление носит условный характер, поскольку в разных участках кожного покрова могут быть обнаружены проявления всех стадий заболевания (пятна, бляшки, папулы, узлы различной величины, некоторые с явлениями некроза). Наиболее частая локализация поражений отмечается на голенях, спине, лице, волосистой части головы
Поражения кожи.Поражение эпидермиса сопровождается зудом, шелушением, гиперкератозом ладоней и подошв, изменением пигментации кожи, алопсцией. В биоптатах пораженных участков кожи имеет место полиморфизм клеточного инфильтрата со значительной примесью зозинофилов и атипичных лимфоидных клеток с мозговидными ядрами, образование микроабсцессов Дарье-Потрие. Картина периферической крови и костного мозга без изменен
Лейкемический вариант грибовидного микоза (синдром Сезари)характеризуется поражением лимфатических узлов, костного мозга и появлением в периферической крови опухолевых лимфоцитов (клеток Сезари).
В гемограмме отмечаются признаки анемии, лейкоцитоз, абсолютный лимфоцитоз, нейтропения
Иммунологический фенотип опухолевых клеток соответствует Т-хелперам (CD2+, CD3+, CD4+, CD5+). •Прогноз заболевания зависит от стадии опухолевого процесса. Средняя продолжительность жизни составляет 5-10 лет. При поражении лимфатических узлов, висцеральных органов, костного мозга прогноз резко ухудшается.
Хронический лимфолейкоз (ХЛЛ) относится к опухолям, первично возникающим в костном мозге в результате опухолевой трансформации чаще В-лимфоцитов, реже Т-лимфоцитов и последующей их моноклональной пролиферацией
ХЛЛ впервые описан в 1845 году, а в середине 60-х гг. охарактеризован Galton и Dameshek как заболевание, сопровождающееся пролиферацией аномальных лимфоцитов. ХЛЛ составляет 30% всех регистрируемых случаев лейкозов в Европе и Америке, значительно реже выявляется в Азии. Частота встречаемости заболевания 2,7-3,0 на 100 тысяч населения. Болеют в основном лица старше 50 лет, мужчины в два раза чаще, чем женщины. В детском и юношеском возрасте заболевание встречается крайне редко. Заболеваемость ХЛЛ наблюдается в 2 раза чаще у представителей белой расы, чем негроидной.
Большинство случаев ХЛЛ составляет В-клеточная форма (95%). На долю Т-клеточной формы приходится около 5% от всех наблюдений ХЛЛ, в основном регистрируемых в странах Азии.
Среди этиологических факторов рассматривается воздействие химических веществ, вирусов. Доказана роль человеческого Т-клеточного вируса I типа (HTLV-I) в развитии Т-клеточного варианта ХЛЛ.
В экспериментальных работах доказана сверхэкспрессия генов семейства Bcl-2 в опухолевых В-лимфоцитах, блокирующих апоптоз, что способствует удлинению продолжительности жизни этих клеток. ХЛЛ относится к медленно прогрессирующим заболеваниям. Опухоль постепенно вытесняет нормальные гемопоэтические клетки, что со временем приводит к развитию недостаточности костномозгового кроветворения
Пролиферация опухолевых В-лимфоцитов в костном мозге, лимфатических узлах, селезенке, реже в других органах (кожа, желудочно-кишечный тракт, почки, легкие и др.) обуславливает клиническую картину заболевания
Клиника.
На протяжении нескольких лет заболевание протекает бессимптомно. Лишь выявление абсолютного лимфоцитоза при исследовании клеточного состава периферической крови может привлечь внимание врача. Заболевание сопровождается общими для многих злокачественных опухолей неспецифическими симптомами - слабостью, быстрой утомляемостью, повышенным потоотделением, потерей массы тела, связанными с опухолевой интоксикацией.
Для больных ХЛЛ характерна повышенная восприимчивость к инфекции вследствие нарушений в системе клеточного и гуморального иммунитета. Наиболее часто встречаются бактериальные и вирусные инфекции со стороны дыхательной и мочевыводящей систем, вызванные стафилококками, стрептококками, различными грамнегативными микроорганизмами. В развернутой стадии заболевания могут наблюдаться инфекции, вызванные Candida и Aspergillus, а также вирусом герпеса и цитомегаловирусом.
Дефект противоопухолевого иммунитета является причиной повышенной склонности больных ХЛЛ к развитию вторичных опухолей. Наиболее часто при ХЛЛ встречается рак кожи и кишечника.
Существуют различные клинические классификации ХЛЛ. В зависимости от локализации опухоли, особенностей течения заболевания в классификации, предложенной А.И. Воробьевым и М.Д. Бриллиант (1985 г., с изменениями и дополнениями 1999 г.), выделено несколько форм ХЛЛ - доброкачественная, прогрессирующая, опухолевая, селезеночная, абдоминальная, костномозговая. Согласно классификации, предложенной Rai К. с соавт., выделяют 5 стадий болезни.
Классификация Binet J. с соавт. предусматривает 3 стадии ХЛЛ, обозначаемые буквами А, В и С, которые коррелируют со средней продолжительностью жизни больных.
A. Помимо предусмотренного диагнозом лимфоцитоза крови и костного мозга, имеется одностороннее или двухстороннее увеличение лимфатических узлов в 1-2 областях, анемия и тромбоцитопеиия отсутствуют.
B. Увеличение лимфатических узлов в 3 областях и более. Анемия и тромбоцитопения отсутствуют.
C. Независимо от количества зон с увеличением лимфатических узлов и наличия увеличения органов имеется анемия и тромбоцитопепия.
В зависимости от стадии заболевания костный мозг может быть нормо- или гиперклеточным. Процент лимфоцитов в стернальном пунктате колеблется в широких пределах от 20-30% вплоть до тотальной мономорфной лимфоидной инфильтрации. По данным трепанобиопсии поражение костного мозга носит очаговый или диффузный характер. Независимо от стадии заболевания диффузная инфильтрация костного мозга лимфоидными клетками сочетается с малой продолжительностью жизни больных (менее 43 мес.) по сравнению с очаговой инфильтрацией (100 мес.).
В начальной стадии заболевания картина периферической крови обычно представлена нормальным или незначительно повышенным количеством лейкоцитов. Как правило, анемия и тромбоцитопения отсутствуют. Основным гематологическим показателем при ХЛЛ является абсолютный лимфоцитоз.
ФАБ-критерии
Согласно рекомендациям экспертов ФАБ группы диагноз ХЛЛ считается установленным при абсолютном количестве лимфоцитов в крови, превышающем 10 Х 103/мкл, наличии более 30% лимфоцитов в костном мозге и иммунологическом подтверждении существования В-клеточного клона лейкемических клеток
В лейкоцитарной формуле процент морфологически зрелых лимфоцитов составляет от 45 до 95%, встречаются единичные пролимфоциты. Имеет место относительная или абсолютная нейтропения. Лимфоциты периферической крови при ХЛЛ характеризуются небольшими размерами (7-10 мкм), округлым ядром, грубым, тяжистым распределением хроматина, отсутствием нуклеол, узкой базофильной цитоплазмой. Встречаются клетки цитолиза.
В развернутой стадии заболевания нарастает лейкоцитоз, относительный и абсолютный лимфоцитоз, нейтропения, наблюдается нормохромная анемия и/или тромбоцитопения
Обнаружение лимфоцитов с расщепленными, перекрученными, не правильной формой ядрами, грубой тяжистой или волокнистой структурой хроматина свидетельствует о возможной лейкемизации лимфосаркомы или Т-клеточном варианте ХЛЛ. В периферической крови могут обнаруживаться единичные миелоциты и метамиелоциты, обычно на фоне инфекционных заболеваний, а также нормобласты
Примерно у 15% больных ХЛЛ наблюдается развитие аутоиммунной гемолитической анемии, реже тромбоцитопении за счет образования аутоантител к эритроцитам или эритрокариоцитам костного мозга, тромбоцитам. Развитие анемии сопровождается раздражением красного ростка в костном мозге, ретикулоцитозом, появлением нормобластов в периферической крови, повышением содержания неконъюгированного (непрямого) билирубина в сыворотке крови. Аутоиммунный генез анемии подтверждается положительной прямой пробой Кумбса и другими тестами, выявляющими аутоантитела
Иммунологический фенотип. В 95% случаев ХЛЛ имеет В-клеточный иммунологический фенотип с экспрессией поверхностных В-клеточных антигенов CD19, CD20. CD24, СП79а и активационных антигенов CD5, CD23. CD43. Экспрессия CD5 считается обязательной для иммунологического подтверждения В-ХЛЛ. Однако описаны редкие наблюдения ХЛЛ (7%) с отсутствием CD5 на В-лимфоцитах. CD23 используют для дифференциальной диагностики В-ХЛЛ и лейкемизации лимфомы из клеток зоны мантии лимфатического узла, имеющей аналогичный В-ХЛЛ иммунофенотип, но без экспрессии CD23. Для ХЛЛ в отличие от нормальных В-лимфоцитов и лимфосарком характерна слабая экспрессия поверхностных иммуноглобулинов (чаще slgM. реже IgM +IgD с одинаковыми легкими цепями
Практически у всех больных наблюдается гипогаммаглобулинемия со снижением концентрации нормальных иммуноглобулинов (IgM, IgG. IgA), что отражает нарушение гуморального иммунитета и повышает чувствительность больных ХЛЛ к инфекциям. При использовании современных методов электрофореза можно обнаружить белок Бенс-Джонса в моче, значительно реже встречается моноклональный иммуноглобулин в сыворотке крови
ЦитогенетикаУ 50-60% больных ХЛЛ обнаруживают клональные хромосомные аберрации, наиболее часто - трисомия 12, структурные дефекты в 13, 14 хромосомах. Изучение онтогенетических особенностей клеток имеет прогностическое значение. Средняя продолжительность жизни больных с хромосомными аномалиями значительно короче (7,7 лет), чем без таковых (до 15 лет).
Прогноз.ХЛЛ является достаточно медленнотекущим заболеванием. Средняя продолжительность жизни составляет около 10 лет.
ИсходНаиболее часто встречается трансформация ХЛЛ в пролимфоцитарный лейкоз, что характеризуется нарастанием лейкоцитоза, числа пролимфоцитов, анемии и тромбоцитопении. Эти изменения сопровождаются резкой лимфаденопатией, спленомегалией, развитием рефрактерности к проводимой терапии.
Читайте также: