
Рефрактерная анемия с кольцевидными сидеробластами
Миелодиспластическим синдромом называют группу гетерогенных клональных заболеваний крови, объединенных следующими признаками: неэффективный гемопоэз, периферическая цитопения, дисплазия в одном или более ростке кроветворения с высоким потенциалом трансформации в острый миелоидный лейкоз.
Недостаточное кроветворение проявляется анемией, повышенной кровоточивостью и подверженностью инфекциям. Миелодиспластический синдром (МДС) встречается у людей любого возраста, в том числе и детского, но в большей степени ему подвержены люди после 60 лет.
По МКБ-10 миелодиспластическим синдромам присваивается код D46.
Причины
Клетки крови синтезируются и созревают главным образом в костном мозге (этот процесс называется миелопоэзом, а ткань, в которой он происходит, называется миелоидной), затем, выполнив свою функцию и состарившись, уничтожаются селезенкой, а на их место приходят новые. При миелодиспластическом синдроме костный мозг теряет способность к воспроизводству клеток крови (всех – эритроцитов, лейкоцитов, тромбоцитов или только некоторых) в необходимом организму количестве, в кровь попадают незрелые клетки (бласты), в результате чего она хуже выполняет свои функции. Это проявляется характерной для МДС симптоматикой. Примерно в 30% случаев процесс миелопоэза становится со временем полностью бесконтрольным, количество бластных форм кровяных клеток увеличивается, вытесняя нормальные, зрелые клетки. Когда количество бластов в крови превышает 20% (ранее пороговым значением было 30%), ставится диагноз острого миелоидного лейкоза.
В зависимости от того, известна ли причина нарушения функции костного мозга, или нет, МДС делится на первичный, или идиопатический, и вторичный. Вторичный возникает в результате угнетения костномозговой функции после химиотерапевтического или лучевого воздействия. Такое воздействие обычно является частью противоопухолевой терапии, т. е. проводится по поводу какого-либо вида рака. В этом случае МДС можно рассматривать как осложнение.
Первичный, или идиопатический МДС возникает спонтанно, без какой-либо предшествующей патологии и по неизвестной причине. Возможно, предрасполагающим фактором является генетический, поскольку при некоторых видах синдрома обнаруживаются хромосомные изменения.
Факторами, способствующими развитию МДС, являются:
- курение;
- контакт с канцерогенными химическими веществами (пестициды, гербициды, бензол);
- воздействие ионизирующей радиации;
- пожилой возраст.
Формы заболевания
Как уже указывалось выше, МДС делится на два вида, первичный и вторичный.
Чаще встречается первичный МДС (около 80% всех случаев), большинство заболевших – пожилые люди (65-75 лет). Вторичным МДС также в основном страдают пожилые люди, по той причине, что и злокачественные опухоли, а значит, и их осложнения, у них встречаются чаще. Вторичный МДС хуже поддается терапии и связан с худшим прогнозом.
Кроме того, МДС делится на клинические типы в зависимости от типа бластных клеток, их количества и наличия хромосомных изменений, эта классификация предложена Всемирной Организацией Здравоохранения (ВОЗ). Согласно классификации ВОЗ, выделяют следующие формы МДС:
- рефрактерная (т. е. устойчивая к классической терапии) анемия;
- рефрактерная цитопения с мультилинейной дисплазией;
- МДС с изолированной делецией 5q;
- МДС неклассифицируемый;
- рефрактерная анемия с кольцевидными сидеробластами;
- Рефрактерная цитопения с мультилинейной дисплазией и кольцевыми сидеробластами;
- рефрактерная анемия с избытком бластов-1;
- рефрактерная анемия с избытком бластов-2.
Стадии заболевания
В протекании МДС выделяют три стадии, которые, однако, не всегда клинически четко отличаются между собой, различия определяются лабораторно. Это стадия анемии, стадия трансформации (промежуточная между анемией и острым лейкозом), и острый миелоидный лейкоз. Не все исследователи согласны с определением острого миелоидного лейкоза как стадии миелодиспластического синдрома, поскольку он относится к миелопролиферативным нарушениям (т. е. тем, которые характеризуются бесконтрольным клеточным ростом), тем самым не полностью соответствуя характеристикам МДС.
Симптомы
Основные симптомы МДС связаны с проявлениями анемии. Пациенты предъявляют жалобы на повышенную утомляемость, приступы головокружения, одышку при физической нагрузке, которая ранее переносилась легко. Анемия связана с нарушением продукции эритроцитов, следствием чего является низкий уровень гемоглобина в крови.
В некоторых случаях развивается геморрагический синдром, который характеризуется повышенной кровоточивостью. Пациент начинает замечать, что даже незначительные поверхностные повреждения вызывают длительно не останавливающееся кровотечение, может появиться кровоточивость десен, частые и спонтанные носовые кровотечения, петехии на коже и слизистых оболочках, а также множественные гематомы (синяки) либо без связи с какой-либо запоминающейся пациенту травмы, либо после незначительного ушиба или даже надавливания. Геморрагический синдром связан с нарушениями тромбоцитопоэза.
У больных с МДС также обнаруживается подверженность инфекционным болезням. Они часто болеют простудными заболеваниями, кожными бактериальными и грибковыми инфекциями. Такое состояние обусловлено нейтропенией (недостаточностью нейтрофилов).
Кроме того, признаками МДС могут быть:
- беспричинное повышение температуры, часто до высоких значений (38 °С и выше);
- снижение веса, уменьшение аппетита;
- гепатомегалия;
- спленомегалия;
- болевой синдром.
В ряде случаев МДС ничем себя не проявляет и обнаруживается случайно во время лабораторного исследования крови по другому поводу.
Диагностика
Основной метод диагностики МДС – лабораторный. При подозрении на миелодисплазию проводятся:
- Клинический анализ крови. При этом обнаруживается анемия (макроцитарная), ретикулоцитопения, лейкопения, нейтропения, при синдроме 5q – тромбоцитоз. Примерно у половины пациентов выявляется панцитопения.
- Биопсия костного мозга. Цитоз обычно в норме или увеличен, но примерно у 10% пациентов он снижен (гипопластический вариант МДС), есть признаки нарушенного гемопоэза одного или нескольких ростков кроветворения, может обнаруживаться повышенное содержание бластных форм, патологических сидеробластов (эритроциты, содержащие отложения железа). Для идентификации аномальных фенотипов проводят исследование иммунофенотипа костномозговых клеток, это позволяет проводить дифференциальную диагностику МДС и неклональных цитопений, что важно для прогноза.
- Цитогенетический анализ. У 40–70 % пациентов обнаруживаются клональные цитогенетические аномалии, особенно часто наблюдается делеция (моносомия) 7 хромосомы (7q), которая является прогностически неблагоприятной.
- Определение уровня железа и феритина в сыворотке. Уровни повышены.
- Определение эндогенного эриропоэтина (при Диагностика МДС проводится лабораторными методами
Для определения МДС разработаны специальные критерии, т. е. условия, при соблюдении которых ставится данный диагноз. Диагностические критерии следующие:
- 1-, 2- или 3-ростковыя периферическая (т. е. обнаруживаемая в периферической крови) цитопения;
- дисплазия: признаки нарушения гемопоэза не менее 10% клеток не менее одного кроветворного ростка;
- характерные цитогенетические изменения (наличие патологического клона).
Цитопения должна быть стабильной и наблюдаться в течение не менее шести месяцев, однако если обнаруживается специфический кариотип, или ей сопутствует дисплазия не менее двух ростков кроветворения, достаточно двух месяцев.
Для постановки диагноза должны быть исключены другие заболевания, сопровождающиеся клеточной дисплазией и цитопенией.
При выявлении цитопении без других признаков МДС диагностируют идиопатическую цитопению, значение которой не установлено; при выявлении дисплазии без цитопении – идиопатическую дисплазию, значение которой не установлено. При этом требуется постоянное наблюдение пациента с повторным исследованием костного мозга через 6 месяцев, поскольку оба этих диагноза способны прогрессировать до МДС и острого миелоидного лейкоза (или другого миелопролиферативного заболевания).
МДС дифференцируется со следующими заболеваниями:
- анемии (прежде всего, мегалобластическая, сидеробластическая и апластическая);
- острый миелоидный лейкоз;
- лейкопения с нейтропенией;
- первичная иммунная тромбоцитопения;
- клональный гемопоэз с неопределенным потенциалом;
- первичный миелофиброз;
- ВИЧ;
- тяжелая интоксикация различной этиологии.
Лечение
В 1997 году была разработана специальная шкала, называемая шкалой IPSS (International Scoring Prognostic System, Международная шкала оценки прогноза), разделяющая пациентов на группы риска. В соответствии с определенной группой риска выбирается лечебная тактика, и, что следует из названия, оценивается прогноз.
Баллы присваиваются с учетом трех факторов:
- количество бластных форм;
- количество пораженных кроветворных ростков;
- цитогенетическая категория.

Рефрактерная анемия относится к одному из самых опасных видов заболеваний крови. В большинстве случаев на фоне патологии развивается острый лейкоз, спасти от которого человека не всегда возможно. Элементы крови при заболевании из костного мозга в кровоток поступают в незрелой форме, из-за чего их нормальное функционирование оказывается невозможным.
Что такое рефракторная анемия

Анемия в мазке крови
Заболевание относится к приобретённым патологиям костного мозга из группы гетерогенных. Кроветворная функция в организме нарушается не только в количественных показателях, но и в качественных. Патология отличается стойкостью к терапевтическому воздействию, которое эффективно против иных видов патологии. Заболевание диагностируется с равной частотой у женщин и мужчин.
При рефрактерной анемии происходит нарушение в выработке ферментных элементов в костном мозге, из-за чего концентрация эритроцитов значительно снижается. При заболевании отмечается резкое падение гемоглобина и понижение количества не только эритроцитов, а также и тромбоцитов и лейкоцитов. Изменения, которые происходят в костном мозге, необратимые. Особая опасность патологии заключается в том, что в течение длительного времени заметной для больного симптоматики не возникает, и о наличии болезни становится известно только в момент острого лейкоза.
Симптомы

Могут иметь место частые беспричинные обмороки
Симптоматика возникает, когда заболевание уже достаточно сильно развито. Первичные симптомы, такие как единичные точечные кровоизлияния под кожей, появление синяков от незначительных ударов и бледность, редко замечаются больным. Когда состояние становится тяжёлым, развиваются следующие признаки болезни:
- слабость, приводящая к непереносимости физических нагрузок,
- частые обмороки, которые происходят без видимых на то причин,
- сильные головокружения,
- регулярные обильные кровотечения из дёсен и носа,
- сильная бледность,
- подверженность инфекционным заболеванием и грибковым поражениям,
- образование синяков даже от простого нажима,
- одышка,
- быстрое исхудание,
- сильные боли в области тазобедренного сустава,
- выраженная дрожь в теле,
- нарушения дыхания,
- боли неопределённого характера,
- нервозность,
- сильная потливость,
- высыпания на коже.
При развитии симптоматики состояние больного ухудшается стремительно, и обойтись без обращения за врачебной помощью в такой ситуации нельзя.
Причины возникновения

Причиной анемии может стать длительный контакт с пестицидами
По причине появления заболевание разделяется на две группы — первичную и вторичную. Первичное нарушение диагностируется у пожилых людей, и причина его развития остаётся пока неизвестной. Вторичная форма нарушения развивается в любом возрасте на фоне длительного проведения радиотерапии или химиотерапии при раке; а также на фоне следующих факторов, вызывающих нарушения в работе костного мозга:
- нахождение в регионе с повышенным радиационным фоном;
- длительное воздействие неблагоприятной экологии;
- длительный и частый контакт с пестицидами, бензином и химическими растворителями;
- наследственные болезни крови;
- длительный приём ряда лекарственных препаратов.
Заболевание в некоторых случаях развивается на фоне приёма противоопухолевых средств, которые могут разрушать не только патологические клетки, но и здоровые. В результате этого нарушается состояние системы кроветворения, из-за чего картина крови резко ухудшается, в значительной степени усугубляя состояние больного.
Диагностика

Жалобы и анамнез дополняются лабораторными тестами
Предположительный диагноз обычно ставится на основании анамнеза и жалоб пациента. Далее для его подтверждения проводится лабораторная диагностика, при которой осуществляются общий анализ крови и исследование тканей костного мозга, полученных при пункции. Также требуется физикальное исследование для оценки тяжести состояния больного.
При изучении тканей костного мозга оценивается их клеточность, соотношение ростков кроветворения, наличие атипичных клеток. Именно данное исследование является основной мерой для диагностики патологии.
Лечение рефрактерной анемии

При выявлении признаков анемии пациенты подлежат лечению
Терапия при заболевании направлена на снижение интенсивности его проявлений, так как вылечить болезнь невозможно. Также при лечении ставится задача не допустить развитие осложнения, которым является острый лейкоз. При выборе терапии учитывается причина появления нарушения, возраст больного и наличие прочих патологий. Основное поддерживающее лечение сводится к следующему:
- внутривенное вливание дефицитных компонентов из состава крови;
- гормональные препараты, стимулирующие образование ростков кроветворения;
- корректировка прочих патологий;
- использование иммуносупрессорных препаратов.
У молодых пациентов при высоком риске появления острого лейкоза проводится курс химиотерапии. Полное выздоровление больного возможно только после операции по пересадке костного мозга, но эффективность метода не 100%.
Осложнения и прогноз

Своевременное лечение помогает достигнуть благоприятных прогнозов
Основное осложнение заболевания — развитие острого лейкоза. Прогноз для больных зависит от стадии патологии и эффективности поддерживающей терапии. При хорошем прогнозе продолжительность жизни составляет до 11 лет, в то время как при плохом больной редко проживает более 6 месяцев. Заболевание относится к категории злокачественных болезней крови.
Профилактика
Эффективные методы профилактики заболевания не разработаны. Единственное, что рекомендуют врачи для снижения риска появления проблем с системой кроветворения, это поддержка иммунитета, полноценное питание и отказ от вредных привычек. Также важно не допускать присутствия факторов, способствующих развитию патологии, и при первых же признаках нарушения обращаться за врачебной помощью.
Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
- Причины и классификация миелодиспластического синдрома
- Симптомы миелодиспластического синдрома
- Диагностика миелодиспластического синдрома
- Лечение и прогноз при миелодиспластическом синдроме
- Цены на лечение
Общие сведения
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.
Дальнейшие исследования позволили выявили у больных с резистентной к терапии анемией, как с увеличением количества бластных клеток в костном мозге, так и без, ряд особенностей. Эти особенности касались в первую очередь морфологии костного мозга (нарушение архитектоники ростков гемопоэза, изменения стромальной ткани, признаки дисплазии кроветворных клеток). Кроме того, при выполнении цитогенетического исследования у таких больных часто выявлялись изменение кариотипа гемопоэтических клеток.
Цитогенетическими и ферментными методами была доказана клональная (опухолевая) природа заболевания. Характерной особеностью клеток, происходящих из опухолевого клона, при миелодиспластическом синдроме является их морфологическая и функциональная неполноценность.
Дальнейшие наблюдения позволили установить, что миелодиспластический синдром является неоднородной группой. У больных миелодиспластическим синдромом отмечались существенные различия в тяжести клинических проявлений, длительности течения заболевания (сроках выживаемости больных), в частоте и скорости озлокачествления заболевания — трансформации в острый лейкоз. Различия в течение заболевания требовали и различной тактики ведения больных, то есть возникла необходимость выделения самостоятельных нозологических форм.
Миелодиспластический синдром (МДС) — это группа заболеваний костного мозга, носящих клональный характер и возникающих в результате мутации стволовой клетки крови. При этом потомки мутировавшей стволовой клетки сохраняют способность к дифференцировке до зрелых клеток. Однако процесс дифференцировки носит неэффективный характер, в результате чего зрелые клетки крови изменены морфологически, уменьшены в количестве и ослаблены в функции.
Сегодня общепринятой является классификация МДС разработанная Франко-Америко-Британской исследовательской группой (FAB) и опубликованная в 1982 году. В основе классификации лежат четыре признака:
— количество бластов в костном мозге;
— количество бластов в периферической крови;
— количество атипичных (кольцевидных) сидеробластов в костном мозге;
— количество моноцитов в периферической крови.
Такой набор признаков позволяет разделить группу схожих заболеваний на самостоятельные нозологические формы (таблица 1), которые отличаются по частоте встречаемости, длительности течения, вероятности озлокачествления (трансформации в острый лейкоз) и требуют различной тактики лечения больных. Частота встречаемости различных заболеваний, выделяемых в группе МДС, длительность выживания больных и вероятность трансформации в острый лейкоз представлены в таблице 2.
Таблица 1. FAB-классификация миелодиспластического синдрома (по Bennett et al., 1982).
а) количество бластов в костном мозге менее 5%
б) количество кольцевых сидеробластов в костном мозге менее 15%
в) количество бластов в периферической крови менее 1%
г) количество моноцитов в периферической крови менее 1х10^9/л
2.Рефрактерная анемия с кольцевыми сидеробластами:
а) количество бластов в костном мозге менее 5%
б) количество кольцевых сидеробластов в костном мозге не менее 15%
в) количество бластов в периферической крови менее 1%
г) количество моноцитов в периферической крови менее 1х10^9/л
3.Рефрактерная анемия с избытком бластов:
а) количество бластов в костном мозге более 5%, но менее 20%
б) количество кольцевых сидеробластов в костном мозге менее 15%
в) количество бластов в периферической крови менее 5%
г) количество моноцитов в периферической крови менее 1х10^9/л
4.Рефрактерная анемия с избытком бластов на стадии трансформации:
а) количество бластов в костном мозге более 20%, но менее 30%
б) количество кольцевых сидеробластов в костном мозге менее 15%
в) количество бластов в периферической крови менее 1%
г) количество моноцитов в периферической крови менее 1х10^9/л
5.Хронический миеломоноцитарный лейкоз:
а) количество бластов в костном мозге менее 20%
б) количество кольцевых сидеробластов в костном мозге любое
в) количество бластов в периферической крови менее 5%
г) количество моноцитов в периферической крови не менее 1х10^9/л
Таблица 2. Частота встречаемости каждого из нозологических вариантов МДС, длительность выживания и вероятность трансформации в острый лейкоз.
частота (%) выживаемость (мес) вероятность(%)
1.Рефрактерная анемия: 25 37 11
2.Рефрактерная анемия с кольцевыми сидеробластами: 18 49 5
3.Рефрактерная анемия с избытком бластов: 28 9 23
4.Рефрактерная анемия с избытком бластов
на стадии трансформации: 12 6 48
5.Хронический миеломоноцитарный лейкоз: 17 22 20
Эпидемиология. МДС — патология старшей возрастной группы. 80% случаев МДС приходится на лиц старше 60 лет. В европейских странах среди лиц 50 — 69 лет регистрируется 40 новых случаев МДС на 1 млн населения, а среди лиц 70 лет и старше — 150 новых случаев на 1 млн населения.
Этиология. Несмотря на многочисленные исследования, причины вызывающие развитие МДС остаются во многом неясными. В группе этиологических факторов рассматривают факторы, способные вызывать мутации клеток и тем самым приводить к развитию опухоли: вирусы, ионизирующее излучение, химические агенты. На сегодняшний день каких-либо этиологических факторов, специфичных для МДС не установлено. В ряде случаев развитию МДС предшествует химиотерапия солидных опухолей.
Патогенез. Отправной точкой в развитии МДС является мутация стволовой клетки крови. Потомки мутировавшей клетки получают биологическое преимущество перед нормальными гемопоэтическими клетками, что позволяет им полностью колонизировать костный мозг, вытесняя нормальные гемопоэтические клетки. Особенностью мутации стволовой клетки крови при МДС является частичное сохранение ее потомками способности к созреванию до зрелых клеток крови. Однако, процесс созревания носит неэффективный характер, что приводит к уменьшению количества зрелых клеток в периферической крови. Кроме количественных изменений в составе клеток периферической крови имеет место и снижение их функциональной активности.
Немаловажную роль в развитии патологического клона гемопоэтических клеток играет стромальное микроокружение, однако конкретные механизмы вовлечение стромальной ткани в патологический процесс при МДС изучены еще недостаточно.
Неэффективный характер гемопоэза (дисплазия кроветворения) имеет хорошо выраженный морфологический эквивалент — изменение как морфологических признаков гемопоэтических клеток, так и их расположения внутри костномозговой полости (изменение архитектоники). Третью составляющую морфологических признаков дисплазии кроветворения образуют изменения стромальной ткани. Основные морфологические признаки дисплазии дисплазии кроветворения представлены в таблице 4 и таблице 5.
Таблица 4. Морфологические признаки дисплазии кроветворения при исследовании аспирата костного мозга (по Bartl R, Frisch B и Baumgart R, 1992).
— большие мегакариоциты с одним или несколькими мелкими круглыми ядрами
— увеличение бластных клеток
— гипо- и гипергранулярность
— базофилия цитоплазмы зрелых клеток
— эозинофилы с кольцевыми ядрами
— моноциты с множественными вытянутыми лопастями цитоплазмы
— афзурофильные гранулы в цитоплазме
Ниже представлены несколько фотографий, иллюстрирующих морфологические признаки дисплазии кроветворения.

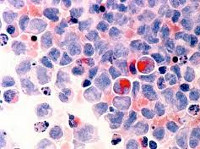
Рисунок 1. Дисплазия эритроидного ростка в костном мозге: мегалобластоидность, асинхронность ядер, тельца Жоли.

Рисунок 2. Дисплазия мегакариоцитарного ростка в костном мозге: микромегакариоцит.

Рисунок 3. Дисплазия гранулоцитарного ростка в костном мозге: значительная редукция числа гранул.

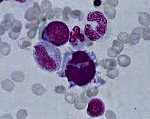
Рисунок 4. Костный мозг больного рефрактерной сидеробластной анемией: кольцевые сидеробласты.
Таблица 5. Гистологические признаки дисплазии кроветворения при исследовании биоптата костного мозга (по Bartl R, Frisch B и Baumgart R, 1992).
Клеточность костного мозга:
— гиперклеточный (свыше 50% случаев)
— нормоклеточный (30-40% случаев)
— гипоклеточный (менее 20% случаев)
— атипичная локализация незрелых предшественников
— атипичная локализация эритроцитарных предшественников
— атипичная локализация мегакариоцитов
— интраваскулярное расположение гемопоэтических клеток
— расширение синусоидов со склерозом стенок
— интростициальный и парамегакариоцитарный фиброз
— увеличение тучных клеток
— увеличение костного преобразования
В неопластическом клоне могут происходить вторичные мутации клеток. В ряде случаев это приводит к развитию в костном мозге клона гемопоэтических клеток, потерявших способность к созреванию в большей степени, чем гемопоэтические клетки из предшествующего клона. Морфологическим эквивалентом этого события является увеличение в костном мозге количества незрелых клеток — бластов. Если количество бластов в цитологическом препарате костного мозга превышает 30%, то говорят о трансформации в острый лейкоз. При этом следует понимать, что данная ситуация не означает развитие нового (второго) заболевания, а является закономерным продолжением течения данной нозологической фомы миелодиспластического синдрома, подчиняющегося закону опухолевой прогрессии. Трансформация в острый лейкоз при МДС эквивалентна развитию бластного криза при хроническом миелолейкозе.
Клиническая картина. Клиническая картина при различных формах МДС схожа и во многом определяется показателями периферической крови. Изменения периферической крови прямо зависят от степени нарушения созревания гемопоэтических клеток. Анемия постоянный и обязательный признак. Для нее характерны гиперхромия (высокий цветовой показатель) и макроцитоз. Уровень снижения гемоглобина может варьировать от умеренного до значительного. От степени и скорости нарастания анемии будет зависеть самочувствие больного. При медленном снижении гемоглобина организм успевает адаптироваться к гипоксии и количество жалоб у больных может быть минимальным. Если анемия развивается быстро, больные предъявляют жалобы на общую слабость, утомляемость, сердцебиение, одышку. Может утяжеляться течение ишемической болезни сердца, появляются признаки сердечной недостаточности.
Снижение количества зрелых гранулоцитов (нейтропения), а также их функциональная несостоятельность влекут за собой инфекционные осложнения. У 10 % больных развиваются стоматиты, гингивиты, пневмонии, инфекция мочевыводящих путей, абсцессы различной локализации, сепсис. У 20 % больных данной группы инфекционные осложнения становятся причиной смерти. Наиболее многочислены осложнения бактериальной природы, возбудителями которых являются Escherichia coli, Pseudomonas pyocyanea, Klebsiella pneumoniae, Staphylococcus aureus и Streptococcus fecalis. Также достаточно часто тяжелые инфекционные осложнения вызываются Pneumocystic carinii, Cryptococcus neoformous, Candida albicaus, Aspergillus fumigatus и цитомегаловирусом, что связано с функциональной неполноценностью Т-лимфоцитов при МДС.
Клинически значимая тромбоцитопения (приводящая к развитию геморрагического диатеза с петехиально-пятнистым типом кровоточивости) встречается у 15 % больных МДС. У половины из них кровотечение или кровоизлияния становятся причиной смерти. В некоторых случаях МДС, как правило у больных рефрактерной анемией, может отмечаться тромбоцитоз. Проявления гиперпластического синдрома в виде спленомегалии, гепатомегалии, лимфоаденопатии и специфического поражения кожи (лейкемиды) имеют место в основном у больных ХММЛ.
Спленомегалия встречается у 17 % таких больных, гепатомегалия у 13 %, а лейкемиды у 10 %.
Диагностика.Отправной точкой диагностического поиска являются, как правило, жалобы связанные со снижением уровня гемоглобина, подкрепляемые выявлением гиперхромной, макроцитарной анемии при исследовании периферической крови. Выявление при первичном осмотре, наряду с анемическими жалобами, явлений геморрагического диатеза и/или гиперпластического синдрома позволяют сформировать представление о
В качестве вспомогательного метода диагностики может быть использовано цитогенетическое исследование кариотипа гемопоэтических клеток. Различные хромосомные поломки выявляются у 48 % больных МДС. Частота аномалий кариотипа варьирует в зависимости от нозологического варианта МДС. Так у больных РА хромосомные поломки обнаруживаются в 30 % случаев, а у больных РАИБтранс в 60 %. Выявление хромосомных аномалий имеет большое значение для определения прогноза течения заболевания.
Наиболее часто встречающимся изменением кариотипа у больных РА является делеция (утрата) части длинного плеча пятой хромосомы (5q-). Данная аномалия чаще выявляется у женщин (соотношение мужчин и женщин среди заболевших составляет 1:5). Для больных с такой хромосомной поломкой характерны ярко выраженые морфологические аномалии мегакариоцитов (микромегакариоциты), тромбоцитоз периферической крови и достаточно благоприятное течение заболевания с низкой частотой трансформации в острый лейкоз.
Диагноз складывается из морфологически подтвержденного представления о наличии у больного миелодиспластического синдрома и окончательно формулируется (нозологическая форма) на основании количественных критериев миелограммы и гемограммы (FAB-классификация):
РА — бласты костного мозга 5%, моноциты периферической крови
Читайте также: