
Рак почек это наследственное

Приглашаем врачей, чья деятельность связана с онкоурологией, к активному сотрудничеству.
Почти каждый десятый случай почечно-клеточного рака имеет наследственный характер. О генетических синдромах, их влиянии на течение болезни и терапию — в системном обзоре на основании последних данных от EAU.
Известны онкологические синдромы, связанные со специфическими мутациями, гистологическим типом опухоли и сопутствующими заболеваниями. Однако ряд наследственных состояний не выдают себя особенностями; заподозрить их можно главным образом по молодому возрасту или семейному анамнезу. Медиана возраста при диагностике наследственного почечно-клеточного рака составляет 37 лет, 70 % опухолей выявляются у пациентов моложе 46 лет. Именно этот возрастной предел повышает вероятность успеха генетического тестирования.
Важное значение имеет анализ наследственных мутаций. Согласно данным Атласа ракового генома (The Cancer Genome Atlas, TCGA), в 6 % случаев светлоклеточного почечно-клеточного рака (сПКР, clear cell renal cell carcinoma — ccRCC) выявляются герминальные мутации, при наличии папиллярного и хромофобного рака их частота равна 9 и 6 % соответственно.
Эта наследуемая по аутосомно-доминантному типу патология характеризуется развитием целого ряда полиморфных опухолей (табл . 1) . Причиной развития синдрома является биаллельная инактивация гена-супрессора опухолевого роста VHL. Поражение почек проявляется наличием кист или светлоклеточного почечно-клеточного рака, обычно мультифокального и билатерального характера VHL-синдром традиционно делится на типы 1 и 2, последний, в свою очередь, — на подтипы (табл 2).
Тактика ведения
Обязательный скрининг пациентов с VHL-отягощенным анамнезом начинается с ретиноскопии в возрасте 5 лет и проведения МРТ головного мозга и брюшной полости каждые 2 года. Необходимы исследование крови и мочи на наличие метанефринов, контроль артериального давления, неврологический осмотр, наблюдение у офтальмолога, отоларинголога и уролога. Генетическая консультация показана пациентам с гемангиобластомами сетчатки глаза и/или ЦНС, лицам молодого возраста и больным с мультифокальными карциномами почек и надпочечников.
Наследуется по аутосомно-доминантному типу. Характеризуется медленным ростом, множественностью, билатеральностью поражения и исключительно папиллярным вариантом 1 типа строения опухоли. В отличие от других синдромов отсутствуют внепочечные проявления. Возраст начала заболевания варьирует; отмечены случаи диагностики болезни у лиц в возрасте 30 лет, к 80 годам фенотипическое проявление приближается к 100 %.
Тактика ведения
Для наблюдения рекомендуются МРТ или КТ с учетом того, что, подобно спорадическому пПКР, наследственные опухоли по большей части гиповаскулярные и при КТ контрастируются лишь на 10-30 единиц Хаунсфилда (HU). На ультразвуковых изображениях поражения часто изоэхогенны по отношению к фоновой паренхиме почки и могут оставаться незамеченными.
Размер опухолевых очагов обычно не превышает 3 см. Выявление активирующих мутаций гена MET привело к испытаниям его ингибиторов. Среди них форетиниб оказался наиболее эффективным у пациентов с герминальными мутациями MET; частичный ответ был получен у половины пациентов с наследственным раком и только в 9 % случаев спорадического пПКР.
Аутосомно-доминантный синдром, характеризующийся поражениями кожи, кистами легких, спонтанным пневмотораксом и многоочаговыми опухолями почек. Причиной синдрома является мутация в гене-супрессоре опухолевого роста FLCN.
У большинства пациентов старше 30 лет развиваются фиброфолликуломы и акрохордоны на лице и верхней части туловища. Двусторонние легочные кисты увеличивают сопутствующий риск пневмоторакса. Примерно у трети пациентов развиваются почечные карциномы различного гистологического строения, большинство из которых имеют промежуточную агрессивность, сравнимую с таковой в случае VHL-синдрома. Опухоли несколько менее агрессивны, чем при других наследственных состояниях, но заболевание не следует рассматривать как доброкачественное, поскольку возможно метастазирование.
Приблизительно 13-34 % пациентов имеют опухоли почек; часто наблюдаются ангиомиолипомы и поликистоз почек По сравнению с болезнью VHL, в рамках которой всегда развивается сПКР, связанные с BHD-синдромом опухолевые образования различаются по гистологическому типу:
- приблизительно 50-67 % опухолей имеют гибридный онкоцитарно- хромофобный тип;
- 23-34 % — хромофобные опухоли;
- 7-9 % — сПКР;
- 3-5 % — онкоцитомы;
- примерно 2 % — папиллярные раки почек.
Тактика ведения
Ежегодное МРТ- или КТ-исследование органов грудной клетки и брюшной полости рекомендовано начинать в возрасте 20 лет. У пациентов без поражения почек МРТ рекомендована каждые 3 года.
Аутосомно-доминантный синдромом, обусловленный мутацией в гене FH. Болезнь характеризуется предрасположенностью к доброкачественному лейомиоматозу кожи и матки, развитием двусторонних папиллярных почечно-клеточных карцином и ранних лейомиосарком матки. В большинстве случаев возникает папиллярный рак 2 типа с градацией ядер по Fuhrman 3-4.
Тактика ведения В качестве скрининга осуществляют:
- ежегодные МРТ-исследования органов брюшной полости;
- наблюдение у гинеколога и дерматолога.
Полиорганный синдром, при котором почти все пациенты имеют ассоциированные кожные проявления, в том числе гипопигментированные пятна, лицевые ангиофибромы и околоногтевые фибромы . У большинства имеются поражения ЦНС (дисплазия коры головного мозга, субэпендимальные узелки и с меньшей частотой — субэпендимальные гигантоклеточные астроцитомы).
Сопутствующие неврологические состояния включают судороги и расстройства аутистического спектра . Другие проявления включают лимфангиолейомиоматоз (ЛАМ) легкого, преимущественно поражающий женщин, миокардиальные рабдомиомы и гамартомы сетчатки.
Рак почки встречается у 1-4 % больных, опухоль часто бывает двусторонней. ПКР классифицируется на 3 типа:
- ТС-связанный папиллярный ПКР;
- Гибридные опухоли (хромофобный рак или онкоцитома);
- Неклассифицированные эпителиальные опухоли.
Тактика ведения
- МРТ органов брюшной полости каждые 1-3 года;
- КТ органов грудной клетки каждые 5-10 лет при отсутствии легочных проявлений;
- МРТ головного мозга;
- Наблюдение у дерматолога, невролога, офтальмолога.
В случаях опухолей высокой жировой плотности или биопсийно доказанных ангиомиолипом с низким содержанием липидов наблюдение является предпочтительным методом до тех пор, пока самая большая опухоль не достигнет в размерах около 4 см, после чего рассматривается селективная ангиоэмболизация . 4-сантиметровый порог основан на исторических данных относительно контроля ангиомиолипом, основанных на более высокой склонности к спонтанному кровотечению.
На основании результатов исследования EXIST-II FDA одобрило эверолимус для лечения ангиомиолипом, связанных с туберозным склерозом.
Это злокачественная эпителиальная опухоль, которая характеризуется развитием параганглиом, феохромоцитом, желудочно-кишечных стромальных опухолей и ПКР. Отличительным гистологическим признаком является обнаружение в цитоплазме атипических клеток вакуолей или хлопьевидных включений, содержащих эозинофильную взвесь.
Распространенность заболевания неизвестна. Небольшая серия случаев показала более ранний возраст начала (средний возраст 30-40 лет, диапазон 15-72 лет) и редкие двусторонние опухоли, которые могут проявлять агрессивное поведение и имеют большой риск развития метастатической болезни.
Таблица 1. Органы-мишени при синдроме Гиппеля—Линдау

Таблица 2. Типы синдрома Гиппеля—Линдау

Тактика ведения
Ведущим методом диагностики является иммуногистохимическое исследование, определяющее потерю экспрессии гена SDH-B. В связи с тем, что что поражение при SDH-дефицитном ПКР является полиорганным, после подтверждения диагноза требуется комплексный междисциплинарный контроль:
- МРТ брюшной полости каждые 2 года;
- анализа мочи и крови на метанефрины;
- анализ на хромогранин А;
- МРТ всего тела (от черепа до таза).
Учитывая возможность метастазирования даже при небольших опухолевых образованиях, ПКР у пациентов с герминальными мутациями гена SDH-B следует лечить аналогично опухолям при синдроме HLRCC. Вероятно, что препараты, подавляющие гликолиз и синтез жирных кислот, также могут использоваться при этих типах рака. В данный момент проводятся клинические испытания вандетаниба в сочетании с метформином.
Наследственный опухолевый синдром, при котором имеется повышенный риск возникновения почечно-клеточного рака. Тип карциномы при этом — светлоклеточный, опухоли могут быть одиночными или множественными, уни- или билатеральными. Это редкая наследственная форма рака почки, достоверно зарегистрированная у 7 семей.
Аутосомно-доминантный синдром, связанный с повышенным риском развития мезотелиомы легких, увеальной меланомы, кожной меланомы, ПКР и, возможно, других злокачественных новообразований.
Взаимосвязь герминальных мутаций гена BAP1 с возникновением злокачественных новообразований была впервые описана в 2011 году, когда исследования выявили повышенную частоту развития меланомы и мезотелиомы. Спустя 2 года было показано, что ПКР является одной из основных опухолей этого синдрома. Однако полный спектр опухолевых поражений, ассоциированных с данным состоянием, является предметом постоянного анализа.
Риск развития ПКР у носителей мутантного гена BAP1 оценивается в 10 %. Из-за небольшого числа зарегистрированных случаев фенотип BAP1-ассоциированного почечно-клеточного рака до сих пор полностью не выяснен. Преимущественно присутствует светлоклеточная карцинома, но есть как минимум 2 опубликованных случая несветлоклеточного рака. Существуют ли носители этой мутации с ранее установленным ПКР, пока неизвестно.
Тактика ведения
Рассматривается аналогично таковой при спорадическом ПКР, хотя зарегистрированы случаи агрессивного течения, поэтому рекомендуются раннее оперативное вмешательство и тщательный скрининг как почечных, так и внепочечных проявлений:
- наблюдение офтальмологом и дерматовенерологом;
- КТ или МРТ органов грудной клетки/брюшной полости.
Является результатом мультигенного наследования (вызывается комбинацией мутаций нескольких генов); каких-либо конкретных генетических аномалии выявлено не было. Гистологически это, как правило, светлоклеточные карциномы, но могут быть и другие варианты. Клиническая картина этих опухолей является типичной и неспецифической.
Тактика ведения
Для пациентов с семейным сПКР рекомендуется проводить генетическое тестирование на VHL, SDH-C, BAP1, TCS1 и TCS2. Если болезнь возникает в раннем возрасте, показано тестирование на SDH- и FH-мутации.
Тактика ведения
Диагностика частично основывается на изучении клинической картины пациента, но наиболее точный результат обеспечивает генетический анализ. Специфического лечения не существует, используется лишь симптоматическая терапия, в том числе и хирургические пособия для удаления новообразований.

Рак — это название, обобщающее целый комплекс смертельно опасных болезней. Заболевания характеризуются тяжелыми опухолевыми процессами и носят злокачественный характер. По данным ВОЗ, ежегодно миллионы людей погибают по причине прогрессирования и осложнений подобных новообразований. При этом заболеть рискует каждый второй.
Одной из основных причин распространенности онкологических заболевания исследователи называют предрасположенность к раку по наследству. Важно учесть, что это свойственно далеко не каждой болезни. Примерно 7% из общего числа разновидностей рака передается по наследству через поколение. Стоит отметить, что это — одни из наиболее агрессивных форм онкологии.
Онкология по наследству: основные факторы
Передается ли рак по наследству? По отношению к некоторым разновидностям злокачественной опухоли ответ будет утвердительным. При наличии родственников, умерших или страдающих от новообразований, целесообразно следить за собственным здоровьем.
Но как понять, передается ли рак по наследству, и по какой линии? Прежде всего следует учесть три основных генетических фактора: диатез, конституцию и врожденную слабость органов. Опираясь на данные, полученные при обследовании, можно определить, расположен ли пациент к раку.
Конституцией называется совокупность индивидуальных особенностей конкретного организма. Фактор формируется на основе наследственности и приобретенных свойств. Конституцию определяют цвет волос и кожи, телосложение, индивидуальные реакции на внешние и внутренние раздражители. Она бывает нормостенической, астенической и гиперстенической.
У каждого типа конституции есть набор предрасположенностей к различным видам аномальных реакций. Данный фактор называется диатезом. С ним неразрывно связана органная слабость — снижение функциональности отдельных органов и тканей.
Передается ли рак по наследству, и по какой линии? Ответ во многом зависит еще и от другого фактора: наличия в геноме заболеваний, передающихся наследственным путем. Например, при альбинизме повышается чувствительность кожных покровов к солнечным излучениям, что может стать причиной опухоли. Наследственные заболевания передаются как по материнской, так и по отцовской линии.
Какой рак передается по наследству?
Передается ли рак по наследству? В случае 7% из общего числа разновидностей злокачественных новообразований это действительно так. Развитие онкологического новообразование обуславливается внешними и наследственными канцерогенными факторами, повышающими активность аномальных клеток.
Но какой рак передается по наследству? Согласно данным исследователей, велика вероятность наследования онкологии следующих органов:
- Желудка или толстого кишечника.
- Молочных желез и яичников.
- Щитовидной железы и органов зрения.
- Кожного покрова
- Легких и почек.
Рассмотрим подробнее каждую из наследственных форм рака.
В случае рака желудка, наследственность распространяется на 10% из общего числа проявлений данного онкологического заболевания. Преимущественно развитие злокачественной опухоли угрожает мужчинам и пациентам со II группой крови.
Помимо рака желудка, наследственность прослеживается и в случаях новообразований толстого кишечника. В 60% опухоль является случайным событием, 10% приходятся на последствия травмы, 30% — онкология по наследству. Как и в случае с раком желудка, у мужчин заболевание встречается чаще. Первые проявления онкологии толстого кишечника проявляются в возрасте 20-30 лет.
Рак молочной железы встречается у женщин чаще, чем другие формы онкологии. О наследственной природе опухоли говорит обнаружение аналогичной болезни у близких родственниц: матери, бабушки, сестры, тети. При наличии в семье пациенток с раком груди риск проявления заболевания у здорового человека возрастает в 3 раза.
В случае, если в семье две и более женщины с данным видом онкологии, вероятность развития опухоли у родственниц возрастает в 5 раз. В особой группе риска находятся лица старше 50 лет.

Еще одним видом рака, который передается по наследству через поколение, является злокачественная опухоль яичников. Составляет примерно 3% от всех типов онкологических заболеваний, поражающих женщин. В случае наличия в семье пациенток с раком яичников, ближайшая родственница (мать, сестра) заболеет с вероятностью в 50%.
Какой рак передается по наследству, если речь идет о щитовидной железе? Специалисты отмечают медуллярное злокачественное новообразования. Фолликулярный и рецидивный папиллярный рак встречается в одной семье крайне редко (менее 1% случаев). Канцерогенным фактором является повышение радиационного фона.
Злокачественная опухоль глаза, ретинобластома, встречается крайне редко. При этом вероятность наследственной передачи высока — 40%. Предрасположенность к раку выявить сложно. Само заболевание проявляется в раннем детстве или во младенческом возрасте.
Онкологическое поражение легкого — одна из самых распространенных форм рака. У мужчин данный тип патологии на первом месте, у женщин — на втором после рака груди. Основной причиной болезни является курение.
Как выяснили исследователи из Оксфордского университета, предрасположенность к раку легкого носит наследственный характер. Лицам, в семьях которых есть пациенты с подобным диагнозом, категорически нельзя курить. Даже одна сигарета в день становится канцерогенным фактором.
Патология почки, возникающая и развивающаяся в детском возрасте – опухоль Вильмса, нефробластома. Семейный формы рака выявляются у 5% из общего числа заболевших. Риск увеличивается, если пациент страдает от наследственных заболеваний мочеполовой системы. Первыми признаками опухолевого процесса являются примеси крови в моче и болевые ощущения в пояснице.
Меланома кожи возникает как при воздействии случайных факторов, так и на основании прямой наследственности. Высок риск развития опухоли при наличии 2 и более больных в одной семье. Канцерогенным фактором является наличие на теле большого количества крупных родинок и пигментных пятен.
Меланома чаще развивается у молодых людей в возрасте от 20 до 40 лет. Наследственный тип опухоли характеризуется множественным очагом поражения: в разных участках тела формируются несколько новообразований. От обычной родинки меланома отличается нечеткими границами, необычным цветом и наличием выделений.
Как избежать передачи онкологии по наследству?
Предрасположенность к раку еще не говорит о том, что вы наверняка заболеете. Наследственность может не проявляться до тех пор, пока канцерогенные факторы не приведут к развитию опухолевого процесса.
Но как избежать рака, который передается по наследству?
Если у вас в семье есть онкобольной, рекомендуется пройти генетическое обследование на предрасположенность к данной форме болезни. В случае положительного результата, рекомендуется отказаться от табакокурения, распития алкогольных напитков, увлечения фаст-фудом и контакта с ядовитыми и канцерогенными веществами. Лучшей профилактикой онкологических заболеваний является здоровый образ жизни.
При наличии угрожающей наследственности, рекомендуется регулярно проходить диагностику особо уязвимых органов. К примеру, женщинам после 40 лет следует не реже 1 раза в год проходить УЗИ молочных желез и осмотр у гинеколога.
Злокачественная опухоль почки формируется в эпителии проксимальных канальцев и собирательных трубочек (почечно-клеточная форма) или в эпителии чашечно-лоханочной системы (переходно-клеточная форма). Среди первичных злокачественных образований у взрослых эта онкопатология занимает 80–85 %.
Виды наследственного рака почки
Выделяют такие наследственные синдромы:
- Аутосомно-доминантный поликистоз почек. Повышает вероятность почечно-клеточной карциномы.
- Наследственная папиллярная карцинома. Это семейный синдром наследственного рака почки, а также высокопенетрантная, аутосомно-доминантная патология.
- Болезнь фон Гиппеля-Линдау. Аутосомно-доминантный синдром, сопровождающийся множественными доброкачественными и злокачественными образованиями, включая светлоклеточную карциному.
- Наследственный рак почек, ассоциированный с зародышевыми мутациями цикла Кребса. Это агрессивные формы почечно-клеточной карциномы, метастазирующие даже при небольшом размере и т. д.
Выявление генетических факторов риска по наследственному раку почки у пациента и его семьи позволяет разработать такую стратегию, которая минимизирует или предотвратит болезнь-ассоциированную заболеваемость. Анализу подлежат такие гены/протеины, как c-MET (локус 7q31), фумаратгидратаза (1q42), фолликулин (17p11), сукцинатдегидрогеназа (5p15), TSC1 (9q3416p13), TSC2 (3p25).
Причины рака почки
Основными факторами риска считаются:
- курение;
- ожирение;
- гипертония;
- мужской пол;
- удаление матки;
- тяжелые болезни почек;
- генетические патологии (вызывают наследственные формы рака почки);
- длительный диализ при хронической почечной недостаточности.
Симптомы рака почки
Основными признаками патологии являются:
- гематурия (наличие крови в моче);
- болевой синдром в проекции почки (боль ноющая, тупая, почечная колика);
- задержка мочи (из-за скопления в мочевом пузыре кровяных сгустков);
- повышение артериального давления (вторичная гипертензия);
- пальпируемое образование в области почки;
- симптомы общего характера (потеря веса, субфебрильная температура, нарастающая слабость).
При метастазах в легкие возникает кровохарканье и боль в груди, при поражении печени — признаки желтухи. Метастазирование в кости сопровождается патологическими переломами, в головной мозг — головными болями, радикулитом, неврологической симптоматикой.
Диагностика рака почки
Для обследования используются такие методы:
- УЗИ;
- КТ;
- рентгенография с внутривенным контрастированием;
- МРТ;
- ангиография;
- сцинтиграфия;
- биопсия опухоли с гистологическим исследованием.
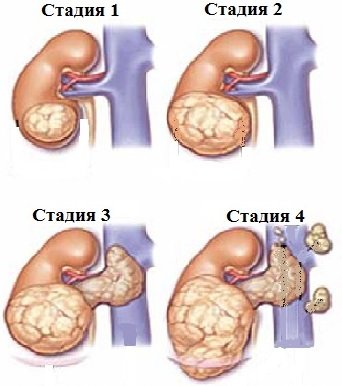
Выделяют четыре стадии рака почки:
- Опухоль ограничена почкой, через капсулу не проникает.
- Образование проникает через капсулу органа.
- Рак почки распространяется на ближайшие лимфоузлы, почечную и нижнюю полую вены.
- Опухоль разрастается на соседние органы (кишечник, поджелудочную железу), появляются метастазы (например, в легких).
Лечение рака почки
Основными методами являются:
Диагностика на ранней стадии онкопатологии позволяет в 90 % случаев добиться излечения.
Наследственные опухолевые синдромы
Наследственный, или семейный, рак почки определяется в 4% всех карцином почки и связан с неопластическими синдромами, для большинства из которых характерны герминальные наследственные мутации в соответствующих генах.
При каждом из синдромов имеется предрасположенность к определенному типу почечной карциномы.
Опухоли нередко множественные, билатеральные, возникают в молодом возрасте.
Наследуется по аутосомно-доминантному принципу, характеризуется развитием капиллярных гемангиобластом ЦНС и сетчатки глаза, светлоклеточного почечноклеточного рака, феохромоцитомы, опухолей поджелудочной железы и внутреннего уха. Причиной развития синдрома служит биаллельная инактивация гена-супрессора опухолевого роста VHL. расположенного на коротком плече хромосомы 3 (3р25-26).
Один аллель этого гена повреждается в результате наследственной герминальной мутации, а другой — теряется уже в опухоли в результате протяженной делеции (потеря гетерозиготности) или повреждается второй мутацией, либо его промоторная область подвергается гиперметилированию. что приводит к инактивации гена VHL.
Поражение почек при VHL-синдроме проявляется наличием кист или светлоклеточного почечноклеточного рака, обычно многоочагового и билатерального. Средний возраст пациентов 37 лет, при этом гемангиобластомы появляются раньше поражения почки.
Данная клиническая картина характерна для болезни 1-го типа. При VHL-синдроме 2-го типа раньше других возникает феохромоцитома надпочечника, а опухоли других локализаций развиваются редко.
Наследуется по аутосомно-доминантному принципу, характеризуется развитием билатеральных почечноклеточных папиллярных карцином типа 1, от микроскопических до клинически значимых по размеру опухолей. В основе рака лежит герминальная активирующая мутация в протоонкогене МЕТ, локализующемся в хромосоме 7q31.
Ген МЕТ кодирует рецептор фактора роста гепатоцитов. Мутации в этом гене при наследственной папиллярной карциноме типа 1 активируют тирозинкиназный домен рецептора, что приводит к стимуляции клетонной пролиферации и инвазивному росту клеток. Диагностика этого наследственного заболевания заключается в идентификации активирующей мутации, которая обычно локализована в экзонах 15-21 гена МЕТ, кодирующих тирозинкиназный домен рецептора.
Синдром BHD проявляется множественными доброкачественными опухолями волосяных фолликулов кожи. У ряда пациентов развиваются эпителиальные опухоли почки, чаще хромофобные и светлоклеточные карциномы, онкоцитомы, кроме того, могут быть кисты в легких, множественные липомы. Клинические симптомы болезни проявляются приблизительно в 50 лет с наличием у пациента в среднем 5 опухолевых очагов.
Причиной синдрома служат мутации в гене-супрессоре опухолевого роста 9HD, локализованном на хромосоме 17 в области 17р11.2. Ген BHD содержит 14 экзонов. которые кодируют белок фолликулин. Все мутации, выявленные на сегодняшний момент, приводят к потере функции белка.
Метанефральные опухоли
Опухоль выявляется у пациентов различных возрастных групп (от 15 мес. до 83 лет), однако наиболее часто встречается в пятой-шестой декаде жизни, преимущественно у женщин. Поражение обычно одностороннее, макроскопически представлено солитарным серовато-желтым узлом, отграниченным псевдокапсулой; средний диаметр 5,5 см (диапазон от 0,3 до 22,0 см). Могут выявляться кальцинаты и небольшие кисты.
При исследовании с малым увеличением может показаться, что опухоль представлена солидным кмпонентом, однако при ближайшем рассмотрении заметны мелкие ацинарные структуры, разделенные прослойками отечного или гиалинизированного матрикса. Клетки могут формировать тубулярные структуры или гнезда, солидные поля, микрокисты, выстланные уплощенными опухолевыми клетками. Часто в опухоли обнаруживаются папиллярные структуры с псаммомными тельцами.
Клетки характеризуются округлым или овоидным ядром немного больше лимфоцита, иногда с центральным расщеплением. Хроматин не конденсирован, ядрышки отсутствуют, фигуры митоза редки или отсутствуют вовсе. Цитоплазма оксифильная. Опухолевые клетки близко расположены и иногда наслаиваются друг на друга (рис. 1 45 и 1.46).

Рис. 1.45. Метанефральная аденома. Тубулярный и солидный компоненты опухоли Окраска гематоксилином и эозином. х100

Рис. 1.46. Метанофральная аденома. Окраска гематоксилином и эозином. х200
При электронной микроскопии видно хорошо развитую базальную пластинку, клеточные контакты, микроворсинки, обращенные в просвет ацинусов. Дифференциальный диагноз следует проводить с солидным вариантом папиллярного почечноклеточного рака и опухолью Вильмса с преобладанием эпителиоидного компонента. Опухоль характеризуется отсутствием экспрессии эпителиального мембранного антиген (ЕМА), рацемазы, цитокератинов (СК7), положительной реакцией с WT1, CD57.
Доброкачественная опухоль, встречающаяся преимущественно у детей от грудного возраста до 15 лет. средний возраст 12 лет. Макроскопически опухоль представлена дольчатым фиброзным узлом в мозговом веществе почки с наличием кист, неровным краем. Опухоль чаще односторонняя, средний диаметр 5 см.
В 1/4 случаев развивается гиперплазия клеток юкстагломерулярного аппарата, что может проявляться стойкой гипертензией. В каждой пятой опухоли можно обнаружить участки хрящевой или глиальной дифференцировки. Иммунофенотип метанефральной стромальной опухоли: CD34+, цитокератины-, S-100-, десмин-.
Редкая опухоль почки, развивающаяся преимущественно у детей (средний возраст 30 мес.). Солитарная опухоль мозгового вещества почки размером от 1,8 до 11,0 см (средний диаметр 3,9 см), с желто-бурой поверхностью разреза, мелкими кистами. Участки некроза или кровоизлияния обычно свидетельствуют о сочетанной опухоли Вильмса.
Опухоль представлена эпителиальным (идентичным метанефральной аденоме) и стромальным (идентичным метанефральной стромальной опухоли) ком понентами. сочетающимися в разных пропорциях.
Митотическая активность эпителиального компонента может варьировать в пределах одной опухоли: наряду с участками с низкой митотической активностью могут встречаться очаги, содержащие более 5 митозов на 20 полей зрения. В опухолях выявляется стойкое диффузное окрашивание СК7 и ЕМА.
Исход заболевания при радикально выполненной операции хороший, независимо от митотической активности.
Мир семимильными шагами двигается к персонифицированной медицине, где важнейшую роль будет играть изучение генетических факторов человека. Это знание поможет не только вылечивать, но и предотвращать страшные болезни, например рак.
Анджелина Джоли – не просто голливудская звезда, но и настоящий боец! Мать и тётя актрисы в относительно молодом возрасте погибли от так называемого наследственного опухолевого синдрома. И Анджелина со свойственной ей решительностью превентивно (то есть заранее) удалила себе молочные железы и яичники (органы – мишени для наследственного рака, высокий риск которого обнаружил её генетический анализ). Первой из публичных личностей она открыто призналась, что сделала операцию, чтобы спастись от ещё несуществующей, но без экстренных мер практически неотвратимой угрозы. Личный риск Джоли, по подсчётам врачей, составлял 87%. Так она создала важный прецедент, который наверняка поможет сохранить жизнь многим женщинам во всём мире!

Это не случайность
Спорадический (то есть случайный) рак может возникнуть у любого. Главная причина болезни – генетические мутации в клетках. С годами в организме их накапливается критическое количество, поэтому развитие опухолей в пожилом возрасте – явление закономерное и почти неотвратимое. Как любят говорить онкологи, каждый человек доживёт до своего рака, если только раньше не погибнет от инфаркта или инсульта.
Но есть и наследственные формы болезни, поколениями поражающие людей из одной семьи. Путь к раку для носителей определённых мутаций в генах короче, чем у других людей. Если обычной клетке для превращения в раковую необходимо накопить 5–6 значимых мутаций, то при наследственном генном дефекте достаточно 4–5.
На долю наследственных опухолевых синдромов приходится лишь 5% среди всех онкологических заболеваний. Но риск стать жертвой страшной болезни для людей, которые носят в себе дефектный ген, не просто выше, чем у других, – он практически фатален. Например, риск заболеть раком молочной железы для обычной женщины составляет около 8–12%, а для носительниц мутаций в гене BRCA1 – больше 60%.
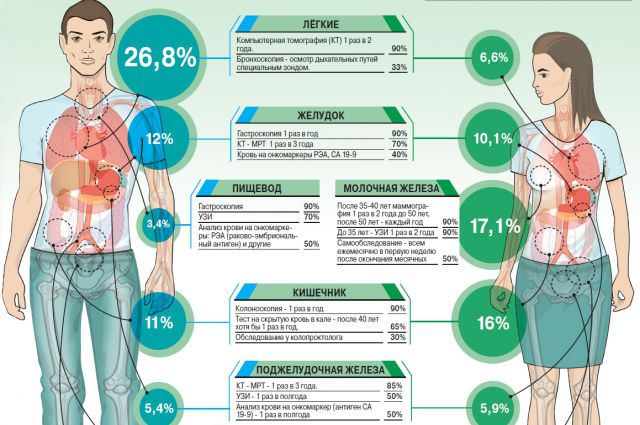
Тревожные признаки
Причина наследственного рака – в полученной от родителей мутации какого-то одного гена, из-за которого опухоль развивается в конкретном органе. Причём наличие семейной истории в этом случае не так важно. Например, если брать самый частый из наследственных синдромов рака молочной железы (РМЖ) и яичников (РЯ), то он может проявиться даже у женщины, в семье которой никто не страдал этим заболеванием. Ведь патогенную мутацию можно унаследовать и от отца.
У наследственного опухолевого синдрома есть чёткие клинические признаки:
Вид наследственного рака
Вклад
в заболеваемость
Гены, мутация в которых повышает риск
Рак молочной железы
BRCA1, BRCA2, CHEK2 и другие
Рак толстой кишки
MLH1, MSH2, MSH6, PMS2
CDH1, BRCA1, BRCA2
Рак поджелудочной железы
Рак предстательной железы
Медуллярный рак щитовидной железы
Приговор отменяется!
Например, при угрозе наследственного рака молочной железы и яичников мероприятия по ранней диагностике (УЗИ, маммография, сдача онкомаркеров) не слишком эффективны. Доказано, что они не гарантируют своевременное выявление опухоли. А рак яичников и вовсе часто выявляется только на запущенных стадиях. Более того, целый ряд новообразований склонен давать метастазы и на первой стадии, поэтому даже раннее выявление заболевания не всегда спасает от смерти. Особенно это характерно для некоторых наследственных опухолевых синдромов. Так, при выявлении РМЖ на первой стадии шансы прожить 5 и более лет есть как минимум у 98% женщин, а для носительниц мутаций в гене BRCA1 этот показатель всего 82%.

Наследственный медуллярный рак щитовидной железы относится к редким заболеваниям и вызывается патогенными мутациями в онкогене RET. Выявление у ещё здорового человека такой мутации – веское основание, чтобы заранее удалить щитовидку. Установлено, что просто частые наблюдения не защитят человека от гибели, а своевременная операция практически полностью снижает риск заболевания. Утрата органа эффективно компенсируется заместительной гормонотерапией.
При угрозе наследственного рака толстой кишки регулярные наблюдения, наоборот, очень эффективны. Исследования доказали, что, если делать колоноскопию каждые 1–2 года, можно достоверно уменьшить риск смерти от этой болезни. Профилактические операции на здоровой толстой кишке обычно не проводятся. Как не удаляют и те органы, утрата которых ведёт к существенному снижению качества жизни. Поэтому органы желудочно-кишечного тракта, части скелета, головной мозг хирурги, конечно, удалять не станут.
Важно
Одно из самых значимых достижений биомедицины последних лет – полногеномное секвенирование (NGS). Оно позволяет всего за несколько дней проанализировать ДНК любого человека. Благодаря ему наши знания о генетических патологиях многократно увеличатся уже в ближайшем будущем. Возможно, полногеномный анализ, который пока ещё слишком дорогое удовольствие (стоит тысячи долларов), в скором времени станет инструментом скрининга.
1. Делайте прививки!
Прививка от гепатита В (вызывает рак печени) и ВПЧ (причина рака шейки матки) может предотвратить 1,1 млн случаев рака в год.
2. Прогресс в онкогематологии
В конце ХХ века 60% детей, больных острым лейкозом, умирали, а сейчас в 90% случаев это заболевание излечивается полностью.
3. Не опускайте руки!
Сейчас в мире насчитывается около 28 млн человек, излечившихся от рака. Большинство из них – женщины, победившие рак груди.
Читайте также: