
Наследственное заболевание характеризующееся доброкачественными опухолями нервов
Нейрофиброматоз — группа наследственных заболеваний, характеризующихся развитием многочисленных нейрофибром, неврином, гемангиом и лимфангиом в подкожной клетчатке. Генетические аспекты
● см. Приложение 2. Наследственные болезни; /сортированные фенотипы
● Множественные нейрофибромы также наблюдают при наследуемых сочетаниях с феохромоцитомой и карциноматозом двенадцатиперстной кишки (162240, R), вариантом синдрома Нунан (601321,R) и при кишечном нейрофибромато-зе (162220, 90.
Нейрофиброматоз I типа (болезнь фон Реклинхаузена)
● Частота 1/3 500. Ген нейрофиброматоза I типа имеет необыкновенно высокую частоту мутаций; половина случаев -спорадические, а не семейные.
● Нейрофибромы (внутрикожные и подкожные) развиваются более чем у 90% пациентов в позднем детстве или юности
● Внутрикожные опухоли, число которых может доходить до 500, проявляют себя как доброкачественные образования, обычно около 1 см в диаметре; иногда они достигают больших размеров (до 25 см), оказывая влияние на физическое состояние пациента
● Подкожные нейрофибромы выглядят как мягкие узелки по ходу периферических нервов.
● Плексиформные нейрофибромы — большие, прорастающие окружающие ткани опухоли, патогномоничные для нейрофиброматоза I типа; вызывают выраженные деформации лица или конечностей; обычно поражают крупные периферические нервы, но иногда могут возникать из черепных нервов или нервов спинного мозга. Основное осложнение -перерождение нейрофибром в нейрофибросаркомы. Нейрофиброматоз I типа сочетается с другими нейрогенными опухолями (например, менингиома, глиома зрительных нервов и феохромоцитома).
● Пятна цвета кофе с молоком. Хотя светло-коричневые родимые пятна на коже наблюдают и у здоровых лиц, более чем 95% лиц, поражённых нейрофиброматозом I типа, имеют 6 и более таких пятен. Размеры пятен более 5 мм до периода половой зрелости и более 1,5 см после полового созревания. Пятна округлой формы, с более длинной осью, ориентированной в направлении хода кожных нервов. Часто отмечают множественные веснушки (лентиго), особенно в подмышечных областях.
● Узелки Лиша — пигментированные узелки на радужке, состоящие из скоплений меланоцитов; наблюдают более чем у 90% лиц с нейрофиброматозом I типа.
● Пороки развития клиновидной кости и истончение коры длинных трубчатых костей, с искривлением и псевдоартрозами большеберцовой кости, кисты костей и сколиоз.
● Лёгкая умственная отсталость.
Нейрофиброматоз II типа — двусторонние невромы слухового нерва, но заболевание можно диагностировать при наличии односторонней опухоли в сочетании с нейрофибромой, менингиомой, глиомой, шванномой. Несмотря на поверхностное сходство между нейрофиброматозом I и И типа, они не являются вариантами одного и того же заболевания. Частота: 1/50 000 населения.
Нейрофиброматоз III типа — ладонные нейрофибромы, бледноватые относительно большие пятна цвета кофе с молоком, двусторонние невромы слухового нерва, менингиомы задней ямки и верхнешейного отдела, спинальные и параспи-нальные нейрофибромы; отсутствие узелков Лиша на радужке; опухоли ЦНС, быстро развивающиеся на втором или третьем десятилетии жизни.
Нейрофиброматоз IV типа — клиника нейрофиброматоза I типа, но без узелков Лиша.
❐ Лечение:
● При множественных периферических нейрофиб-ромах хирургическое лечение неэффективно
при нейрофиброматозе II и III типов — хирургическое иссечение с последующей лучевой и химиотерапией.
❐ Синонимы
● Ней-ромезодерматодистрофия См. также рис. 3-20
✎ МКБ: Q85.0 Нейрофиброматоз (незлокачественный) МШ
Доброкачественные новообразования (опухоли) центральной нервной системы образуются, когда здоровые клетки головного или спинного мозга начинают расти бесконтрольно, образуя объемную опухолевую массу. Доброкачественные опухоли могут расти, причем достаточно интенсивно, но не образуют метастазы и не опасны для жизни больного при своевременном удалении или лечении.
Доброкачественные опухоли центральной нервной системы – виды, признаки, причины
Опухоли ЦНС особенно проблематичны для лечения, поскольку активно влияют (даже при небольших размерах) на мыслительный процесс, координацию, восприятие и все пять органов чувств.
Мозг — центральное звено нервной системы, отвечающее за мыслительный процесс, эмоции и память. Он контролирует все пять чувств: осязание, обоняние, слух, зрение и вкусовые ощущения. Также его задачей является обеспечение движения и других основных функций организма, таких как сердцебиение, кровообращение, дыхание и прочие. Спинной мозг состоит из нервов, передающих нервные импульсы от головного мозга к конечностям и всему телу. Опухоли с низкой степенью опасности принято считать доброкачественными. Признаки доброкачественного новообразования ЦНС:
- медленный рост;
- низкая вероятность рецидива после удаления;
- отсутствие метастазов;
- удаление при помощи обычной хирургической процедуры (без необходимости химиотерапии или облучения).
- вирусные и воспалительные заболевания ЦНС;
- наследственность;
- неблагоприятные экологические условия.
Доброкачественные новообразования головного мозга
Несмотря на то, что доброкачественные опухоли не причиняют большого вреда здоровью и после удаления редко появляются снова, после их обнаружения пациент должен находиться под постоянным наблюдением специалиста.
Далее — подробнее об основных видах опухолей головного мозга.
1. Глиомы (разделяются на астроцитомы, олигодендроглиомы и эпендимомы).
Иногда глиома может представлять собой смешанную опухоль, то есть содержать несколько различных типов клеток. Важное значение имеет место расположения опухоли в головном мозге. Стволовые мозговые глиомы тяжело лечить, независимо от статуса злокачественности. Ствол мозга — деликатная часть этого важного органа, и полное удаление опухоли с этого участка практически невозможно.
Астроцитомы наиболее распространенный тип опухолей мозга, как у взрослых, так и у детей. Астроциты — это клетки головного мозга, поддерживающие работы нейронов. Астроцитомы могут быть быстро растущими и фокусными, то есть с четкой границей между опухолью и здоровой тканью мозга. Диффузные астроцитомы не имеют четких границ.
Олигодендроглиомы развиваются из клеток-олигодендроцитов. Эти клетки производят вещество миелин, покрывающее нервы в головном мозге. Миелин помогает нервным импульсам быстрее перемещаться по нервам. Олигодендроглиомы образуются в височных и лобных долях мозга.
2. Менингиомы — опухоли оболочки мозга.
Поражают головной мозг и мозжечок, преимущественно доброкачественные, удаляются единоразово хирургическим путем, не рецидивируют. Исключение составляет атипичная менингиома, которая имеет высокий риск повторного роста.
3. Гемангиобластомы — новообразования клеток кровеносных сосудов мозга.
Растут медленно, не образуют метастазов, но удаляются крайне тяжело. Иногда являются проявлением редкого синдрома Гиппеля-Линдау, передающегося по наследству.
4. Краниофарингиомы — редкие доброкачественные опухоли, образующиеся у основания мозга, чуть выше гипофиза.
Быстро растут, вызывают проблемы со зрением, гормональные изменения. Провоцируют прибавку в весе и проблемы с ростом.
5. Лимфомы.
Церебральные лимфомы, или лимфомы ЦНС. Редко бывают доброкачественными.
6. Герминогенные опухоли (половых клеток).
Место образования: гипофиз или шишковидная железа.
7. Гипофизарные опухоли (аденомы, гиперплазия).
Также называются нейроэндокринными опухолями. Приводят к проблемам с ростом, половым созреванием, гормональным нарушениям.
8. Опухоль шишковидной железы.
Пинеальная железа располагается в центральной части головного мозга, позади верхней части ствола. Она вырабатывает вещество мелатонин. Шишковидные опухоли встречаются редко (около 1-2 случаев на 100). Дополнительные названия: пинеоцитомы и пинеобластомы. Вызывают головные боли, блокируют циркуляцию физиологических жидкостей в головном мозге, нарушают нормальное движение глаз, приводят к проблемам со зрением.
9. Медуллобластомы (примитивные нейроэктодермальные опухоли).
Место локализации: мозжечок, задний мозг. Имеют высокую степень вероятности перерождения в злокачественную опухоль, быстро растут.
10. Гемангиоперицитомы.
Образуются из клеток-перицитов, содержащихся в капиллярах и сосудах головного мозга. Являются типом саркомы и быстро растут, часто злокачественны, образуют метастазы.
Доброкачественные новообразования спинного мозга
Образуются редко, наиболее распространенными типами доброкачественных новообразований спинного мозга является астроцитома, эпендимома и нейрофиброма.
Астроцитома
Астроцитома спинного мозга – доброкачественное, медленно растущее новообразование спинного мозга. Представляет собой частично кистозную, то есть заполненную жидкостью, и частично твердую опухоль. Астроцитомы удаляются хирургическим путем, редко рецидивируют. В зависимости от области появления вызывают такие симптомы, как головокружение, потерю равновесия, головные боли, обмороки, нарушения зрения, проблемы со слухом и другие неврологические симптомы.
Эпендимома
Доброкачественная опухоль с четкой границей между опухолевой областью и здоровой частью спинного мозга. Несмотря на доброкачественность, могут появляться после удаления, особенно, если удалены не полностью. Папиллярная эпендимома — одна из разновидностей эпендимомы классической, образуется в нижней части спинного мозга. Это мягкая, легко фрагментированная опухоль, которую легко удалить.
Нейрофиброма
Доброкачественные опухоли, возникающие на внешней оболочке нервных корешков. Растут медленно, однако приводят к нарушению координации, снижению чувствительности кожи, онемению конечностей и снижению контроля мочевого пузыря.
В целом опухоли спинного мозга разделяются на экстрамедуллярные и интрамедуллярные. Экстрамедуллярные опухоли развиваются в опорной сети клеток, окружающих спинной мозг и не вторгаются внутрь. Однако они оказывают компрессию на спинной мозг, нарушая его функции. Примером экстрамедуллярной опухоли является нейрофиброма, менингиома.
Интрамедуллярные опухоли образуются из клеток внутри спинного мозга. Пример интрамедуллярной опухоли – это астроцитома и эпендимома.
Доброкачественные новообразования черепных нервов
Акустические невриномы (вестибулярные шванномы) — это доброкачественные новообразования черепных нервов. Их дополнительное название: опухоли мостомозжечкового угла.
Симптомы акустических неврином
Тугоухость сенсорного происхождения, шум в ушах, потеря равновесия, головные боли, полная потеря слуха, звон и шум в ушах, головокружение. Невриномы и шванномы часто являются признаком нейрофиброматоза, наследственного заболевания (болезни фон Реклингхаузена).
Лечение акустических неврином
Акустические невриномы не вторгаются и не разрушают ткани, как это часто бывает с раковыми опухолями. Однако если их не лечить, они пережимают стволы мозжечка, соединяющие мозжечок и головной мозг, и приводят к неврологическим нарушениям. Показано хирургическое удаление.
Первая жертва бубонной чумы: в Монголии скончался подросток
24.1. Нервно-мышечные заболевания
Наследственные нервно-мышечные заболевания – большая гетерогенная группа болезней, в основе которых лежит генетически детерминированное поражение нервно-мышечного аппарата. Заболевания характеризуются мышечной слабостью, мышечными атрофиями, нарушениями статических и локомоторных функций.
При постановке диагноза учитываются возраст проявления первых клинических симптомов заболевания, локализация атрофии и характер распространения миодистрофического процесса (восходящий, нисходящий, наличие или отсутствие псевдогипертрофий, фасцикуляций, расстройств чувствительности, пароксизмов мышечной слабости), а также темп течения.
Прогрессирующие мышечные дистрофии – наиболее обширная группа. В зависимости от характера первичных изменений условно различают первичные (миопатии) и вторичные формы прогрессирующих мышечных дистрофий (денервационные амиотрофии – спинальные и невральные).
Наследственные пароксизмальные миоплегии – группа нервно-мышечных заболеваний, характеризующихся внезапными приступами мышечной слабости и плегиями. Наиболее распространенными из наследственных пароксизмальных миоплегии являются гипо-, гипер– и нормокалиемическая формы. Патогенез неясен. Предполагается генетически детерминированный дефект мембраны сарколеммы, нарушающий проницаемость для ионов натрия и калия,
Гипокалиемическая форма пароксизмальной миоплегии (болезнь Вестфаля). Заболевание описано Вестфалем в 1895 г. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 6—15 лет. Пароксизмы характеризуются внезапным в ночные или утренние часы развитием мышечной слабости, обездвиженности, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами – лабильностью пульса, артериального давления, гипергидрозом. Приступы бывают парциальными, охватывающими небольшую группу мышц, и генерализованными. Во время приступа возникают нарушения сердечно-сосудистой деятельности: систолический шум, изменения ЭКГ. Сознание всегда сохранено. Средняя продолжительность приступа – несколько часов, крайне редко пароксизмы держатся несколько суток. Содержание калия в крови во время приступа менее 2 ммоль/л и ниже. Частота приступов вариабельна. Они провоцируются перееданием пищи, богатой углеводами, охлаждением, физическими нагрузками.
Лечение. Диета, богатая калием (чернослив, курага, картофель, изюм). Для купирования приступа назначают 10% раствор хлорида калия внутрь (по 1 столовой ложке каждый час) или 0,5% раствор в изотоническом растворе хлорида натрия внутривенно (2—2,5 г на 500 мл раствора в течение часа). Целесообразно применять также панангин внутривенно капельно.
Гиперкалиемическая форма пароксизмальной миоплегин (болезнь Гамсторп). Заболевание описано И.Гамсторп в 1956 г. Наследуется по аутссомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 1—5 лет. Симптоматика сходна с пароксизмами при гипокалиемической форме и характеризуется внезапным развитием мышечной слабости, плегиями, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами. В отличие от гипокалиемического гиперкалиемический паралич развивается обычно днем, сопровождается выраженными парестезиями, сочетается со слабостью мышц лица, артикуляционного аппарата, имеет меньшую продолжительность (30—40 мин). Во время приступа содержание калия в крови повышается до 6—7 ммоль/л. Частота приступов вариабельна: от ежедневных до нескольких раз в месяц. В межприступные периоды неврологическая симптоматика отсутствует. Провоцирующими факторами являются голодание, физические нагрузки, вызывающие утомление.
Лечение. Диета с повышенным содержанием углеводов, поваренной соли, ограниченным количеством калия. Вводят 40 мл 40% раствора глюкозы внутривенно вместе с инсулином подкожно; 20 мл 10% раствора хлорида кальция внутривенно.
Нормокалиемический (периодический) паралич. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется до 10-летнего возраста. Особенностью ее является сравнительно медленно (в течение нескольких суток) пароксизмально нарастающая умеренная слабость в мышцах туловища, конечностей и в жевательной мускулатуре, а также медленный (1—2 нед) регресс симптоматики. Провоцирующими факторами являются продолжительный сон, длительное пребывание в одной позе, переохлаждение.
Лечение. Диета, богатая поваренной солью. Назначают ацетазоламид (диакарб).
Течение. Все формы пароксизмальных миоплегий медленно прогрессируют. Прогноз при своевременно поставленном диагнозе, проведении экстренных мероприятий и дифференцированной медикаментозной терапии благоприятный.
Диагностика и дифференциальный диагноз. Диагноз строится на основании генеалогического анализа, особенностей клинической картины, с учетом возраста, в котором начинается заболевание, времени возникновения пароксизма (ночью, утром, днем, в неопределенное время), степени выраженности мышечной слабости, частоты и длительности приступа, провоцирующих факторов, данных лабораторного биохимического исследования (содержание биоэлектрической активности мышц).
Дифференцировать заболевание следует от миоплегий, развивающихся в результате первичных эндокринных заболеваний, – тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
Лечение. Показана диета, богатая поваренной солью. Назначают диакарб.
24.2. Пирамидные и экстрапирамидные дегенерации
Хроническое прогрессирующее заболевание нервной системы, клинически проявляющееся изменениями мышечного тонуса и непроизвольными тоническими сокращениями мышц туловища и конечностей.
Этиология и патогенез. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. Тип наследования при идиопатической торсионной дистонии как аутосомно-доминантный, так и аутосомно-рецессивный. Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Имеются указания, что в патогенезе наследственной торсионной дистонии имеет значение нарушение допаминового обмена. При обследовании у этих больных обнаруживается повышение содержания допамин-?-гидроксилазы в сыворотке крови.
Патоморфология. Дистрофические изменения обнаруживаются преимущественно в мелких нейронах в области скорлупы чечевицеобразного ядра, реже – в других базальных ганглиях.
Клинические проявления. Развивается заболевание постепенно, в 2/3 случаев в возрасте до 15 лет. В детском возрасте первыми симптомами болезни могут быть нарушение походки, спастическая кривошея; у взрослых чаще встречаются первично-генерализованные формы. В результате нарушения соотношения функции мышц-синергистов и антагонистов возникают насильственные длительные тонические сокращения мышц туловища, головы, тазового пояса, конечностей, обычно ротаторного характера, сочетающиеся с атетоидными движениями в пальцах. Создается впечатление, что мышцы постоянно сокращаются для преодоления действия антагонистов. Возникающие позы, даже самые неудобные, сохраняются в течение длительного времени. Гиперкинезы усиливаются при волнении, активных движениях, во сне исчезают. Постепенно, по мере прогрессирования заболевания, поза пациента становится постоянно дистонической, с усиленным поясничным лордозом, флексией бедер, медиальной ротацией рук и ног. В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. При локальных дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения и возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта и непроизвольные движения языка), блефароспазм, щечно-лицевая, щечно-язычная дистония, хореоатетоз.
Течение и прогноз. Заболевание в большинстве случаев неуклонно прогрессирует. Иногда отмечаются различной длительности ремиссии. Быстро происходит глубокая инвалидизация больных и наступает летальный исход, особенно при генерализованной форме.
Лечение. Длительное, симптоматическое. Применяют комбинации холинолитиков и седативных препаратов, в некоторых случаях эффективно использование леводопы. Назначается также галоперидол или резерпин. Очень редко прибегают к стереотаксическим операциям на подкорковых ядрах.
Семейная атаксия Фридрейха – наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых канатиков спинного мозга. Тип наследования аутосомно-рецессивный, с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
Патоморфология. Обнаруживаются дегенеративные изменения в проводящих путях задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени – Бурдаха, Флексига, Говерса, волокнах пирамидного пути, задних корешках, а также в клетках коры мозжечка, подкорковых ганглиев, коры большого мозга.
Клинические проявления. Начало заболевания относится к 6—15-летнему возрасту. Первым симптомом болезни является неустойчивая походка, которая была охарактеризована Шарко как табетически-мозжечковая. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на верхние конечности и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Меняется почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередки патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен.
Заболевание медленно прогрессирует. Средняя продолжительность жизни 10—15 лет с момента его развития.
Диагностика и дифференциальный диагноз. Заболевание распознается на основании характерных симптомов – деформаций стоп по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы и флексия концевых фаланг), поражения миокарда, эндокринных расстройств.
Дифференцировать заболевание следует от церебрального сифилиса, рассеянного склероза, фуникулярного миелоза и других форм мозжечковых дегенерации.
Лечение. Применяются симптоматические средства: общеукрепляющие препараты, лечебная физкультура, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Мозжечковая атаксия Пьера Мари – наследственное дегенеративное заболевание с преимущественным поражением мозжечка и его проводящих путей. Тип наследования аутосомно-доминантный. Возникает заболевание в возрасте 20 лет и старше.
Патоморфология. Выявляется дегенеративное поражение клеток коры и ядер мозжечка, спиноцеребеллярных путей в боковых канатиках спинного мозга, в ядрах моста и продолговатого мозга.
Клинические проявления. Заболевание проявляется нарушениями функций мозжечка и его связей. Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые симптомы сочетаются с умеренными или выраженными признаками пирамидной недостаточности (повышение сухожильных и периостальных рефлексов, клонусы стоп), а иногда с глазодвигательными нарушениями (косоглазие, птоз, недостаточность конвергенции). Характерным признаком является в различной степени выраженное снижение интеллекта.
Диагностика и дифференциальный диагноз. Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии Пьера Мари и атаксии Фридрейха. Нужно учитывать тип наследования заболевания, возраст, в котором развиваются первые симптомы, характер изменения сухожильных рефлексов (при атаксии Фридрейха они снижены), наличие зрительных и глазодвигательных расстройств при атаксии Пьера Мари, деформации стоп и скелета. Рассеянный склероз в отличие от семейной атаксии Пьера Мари характеризуется ремитирующим течением, большей выраженностью нижнего спастического парапареза, расстройством функций тазовых органов.
Лечение. Симптоматическое.
Группа наследственных заболеваний нервной системы, характеризующихся дегенеративными изменениями нейронов мозжечка, ядер нижних олив и моста мозга, в ряде случаев – ядер черепных нервов каудальной группы, в меньшей степени – поражением проводящих путей и клеток передних рогов спинного мозга, базальных ганглиев. Заболевания отличаются типом наследования и различным сочетанием клинических симптомов. По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенерации.
Тип I – оливопонтоцеребеллярная дегенерация Менделя.Наследуется по аутосомно-доминантному типу. Течение медленно прогрессирующее. Проявляться может в возрасте от 11 до 60 лет. Клиническая картина складывается из симптомов поражения мозжечка (атаксия, мышечная гипотония, скандированная речь с элементами дизартрии, интенционное дрожание), ядер каудальных черепных нервов (дизартрия, дисфагия), подкорковых ганглиев (гиперкинезы); реже выявляются пирамидные и глазодвигательные симптомы.
Тип II – оливопонтоцеребеллярная дегенерация Фиклера—Винклера.Наследуется по аутосомно-рецессивному типу. Проявляется в возрасте от 20 до 80 лет симптомами поражения мозжечка, преимущественно атаксией в конечностях. Чувствительность и сухожильные рефлексы не изменены. Парезов не наблюдается.
Тип III – оливопонтоцеребеллярная дегенерация с ретинальной дегенерацией.Наследуется по аутосомно-доминантному типу. Возникает в молодом возрасте. Наряду с мозжечковыми и экстрапирамидными симптомами определяется прогрессирующее снижение остроты зрения вследствие пигментной дегенерации ганглиозных клеток сетчатки.
Тип IV – оливопонтоцеребеллярная дегенерация Шута—Хайкмана.Наследуется по аутосомно-доминантному типу. Проявляется в детском и молодом возрасте. Кроме мозжечковых симптомов, выявляется поражение ядер VII, IX, Х и XII пар черепных нервов (паралич лицевого нерва, бульбарные симптомы) и задних канатиков спинного мозга (расстройства мышечно-суставного чувства и вибрационной чувствительности).
Тип V – оливопонтоцеребеллярная дегенерация с деменцией, офтальмоплегией и экстрапирамидными нарушениями. Тип наследования аутосомно-доминантный. Развивается в среднем возрасте. Характеризуется деменцией, прогрессирующей офтальмоплегией, экстрапирамидными и мозжечковыми симптомами.
Дифференцировать оливопонтоцеребеллярные дегенерации следует от наследственной атаксии Фридрейха и Пьера Мари, прогрессирующих форм рассеянного склероза, опухолей мозжечка, ювенильных форм паркинсонизма.
Лечение. Симптоматическое. Проводят курсы неспецифического общеукрепляющего лечения, массаж, лечебную физкультуру.
Опухоли периферических нервов — опухолевые новообразования, поражающие ствол или оболочки нервов периферической нервной системы. Клинически проявляются парестезиями, болью, нарушением функции нерва (онемением и мышечной слабостью в зоне его иннервации). Диагностика включает клинический и неврологический осмотр, УЗИ, МРТ, электрофизиологические исследования. По показаниям проводится вылущивание опухоли или ее удаление вместе с участком нерва. При выявлении злокачественного характера новообразования оно удаляется вместе с нервным стволом в пределах интактных тканей.
- Причины опухолей периферических нервов
- Симптомы опухолей периферических нервов
- Основные виды опухолей периферических нервов
- Диагностика опухолей периферических нервов
- Лечение опухолей периферических нервов
- Цены на лечение
Общие сведения
Опухоли периферических нервов — достаточно редкая патология нервной системы. Встречаются у лиц любого возраста, чаще у взрослых. Наиболее распространены опухоли серединного, локтевого, бедренного и малоберцового нервов. В неврологии и онконейрохирургии основополагающим является разделение новообразований периферических нервных стволов на доброкачественные и злокачественные. Доброкачественными являются нейрофиброма, невринома (шваннома), перинейрома, злокачественными — нейрогенная саркома (злокачественная шваннома). Нейрофибромы зачастую носят множественный характер. В 50% случаев они ассоциированы с нейрофиброматозом Реклингхаузена. В отдельных случаях доброкачественные опухоли периферических нервов могут брать начало в жировых клетках (липомы) и в сосудах (ангиомы) эпиневрия. От опухолевых образований нервных стволов следует отличать интраневральный ганглий (ганглион, псевдоопухолевую кисту) нерва, представляющий собой внутриневральное скопление муцинозной жидкости, заключенное в плотную оболочку.
Периферические нервные стволы состоят из нервных волокон, каждое из которых покрыто слоем шванновских клеток. Нервные волокна внутри периферического нерва сгруппированы в отдельные пучки, окруженные соединительнотканной оболочкой - периневрием. Между пучками находится эпиневрий, представляющий собой рыхлую соединительнотканную структуру с расположенными в ней сосудами и скоплениями жировых клеток. Снаружи нервный ствол покрывает эпиневральная оболочка. Анатомически нервные волокна представляют собой отростки нейронов, расположенных в спинном мозге или нервных ганглиях. Сами нейроны в процессе своего развития утрачивают способность к восстановлению, но их отростки способны регенерировать. Таким образом, если тело нейрона сохранно и нервное волокно не имеет препятствий для роста, оно способно восстановиться.
В состав одного периферического нерва входят как безмиелиновые, так и миелиновые (мякотные) волокна. Последние имеют т. н. миелиновую оболочку, образованную слоями миелина, многослойно обворачивающими нервное волокно подобно рулону. Основная функция волокон — это проведение нервных импульсов, аналогично тому, как электрический ток движется по проводам. По миелиновым волокнам импульс проходит в 2-4 раза быстрее, чем по безмиелиновым. Если импульсы идут от центра к периферии, то такое волокно носит название эфферентное (двигательное), если импульсы проходят в обратную сторону, то волокно называется афферентное (чувствительное). Нервные стволы могут состоять только из афферентных или только из эфферентных волокон, но чаще они бывают смешанными.
Причины опухолей периферических нервов
Новообразования периферических нервных стволов возникают как результат бесконтрольного деления клеток различных тканей, входящих в структуру нерва. Так, невринома берет свое начало в шванновских клетках оболочки нервных волокон, нейрофиброма — в клетках соединительной ткани эпи- и периневрия, липома — в жировых клетках эпиневрия. Причины опухолевой трансформации нормально функционирующих клеток нервных стволов точно не известны. Предполагают онкогенное влияние радиации и хронического воздействия некоторых химических соединений, а также загрязненности окружающей среды. Ряд исследователей указывает на роль биологического фактора — онкогенного воздействия на организм отдельных вирусов. Кроме того, имеет значение пониженный фон противоопухолевой защиты организма. Провоцирующим триггером может выступать повреждение нерва вследствие травмы. Нельзя исключить и наследственно детерминированную предрасположенность к возникновению опухолей. Установлено, что у пациентов с нейрофибромами и болезнью Реклингхаузена имеются генные мутации в хромосоме 22, обуславливающие недостаточность фактора, ингибирующего опухолевую трансформацию шванновских клеток.
Симптомы опухолей периферических нервов
На ранних стадиях своего развития опухоли периферических нервов обычно имеют субклиническое течение. Типично медленное развитие опухолевого процесса. При поражении мелких ответвлений нервных стволов клинические проявления могут вообще отсутствовать.
Злокачественные опухоли периферических нервов характеризуются интенсивным болевым синдромом, усиливающимся при перкуссии и пальпации по ходу нерва. В зоне иннервации пораженного злокачественной опухолью нервного ствола отмечается неврологический дефицит — снижение силы иннервируемых мышц (парез), гипестезия (чувство онемения и сниженная болевая чувствительность кожи), трофические изменения (бледность, истончение, похолодание, повышенная ранимость кожи). При злокачественном характере новообразования нерва оно плотно спаяно с прилежащими тканями и не смещается относительно них.
Нейрофиброма — доброкачественное новообразование, развивающееся из фиброцитов соединительнотканных структур нервного ствола. Опухоль может быть единичной или множественной (чаще при нейрофиброматозе). Плексиформные нейрофибромы широко инфильтрируют ствол нерва и прилежащие ткани. Такие опухоли наиболее часто встречаются при болезни Реклингхаузена 1-го типа. В 5% случаев плексиформная нейрофиброма претерпевает злокачественную трансформацию.
Невринома (шваннома) — новообразование, берущее начало из шванновских клеток нерва. Чаще наблюдается в возрастном периоде от 30 до 60 лет. Представляет собой округлое утолщение нервного ствола. Обычно имеет единичный характер и медленный рост. Крайне редко отмечается малигнизация с возникновением нейрогенной саркомы.
Перинейрома (периневрома) — редкая доброкачественная опухоль из клеток периневрия. Имеет вид единичного или мультифокального утолщения нерва, протяженностью до 10 см.
Липома — не нейрогенная доброкачественная опухоль, развивающаяся из жировой ткани эпиневрия. Чаще встречается среди лиц, страдающих ожирением. Имеет желтую окраску и плотно срастается с нервным стволом. Не малигнизируется.
Нейрогенная саркома (нейрофибросаркома, злокачественная шваннома) — злокачественная опухоль из оболочек нерва, составляет 6,7% от общего числа сарком мягких тканей. По микроскопическому строению схожа с фибросаркомой. Более подвержены заболеванию лица мужского пола и среднего возраста. Нейрогенная саркома локализуется преимущественно на периферических нервах конечностей, реже — на шее. Редко метастазирует (примерно в 12-15% случаев). Наиболее типичны метастазы в легкие и лимфатические узлы. По микроскопическим особенностям выделяют железистый, меланоцитарный, эпителиоидный вариант опухоли.
Диагностика опухолей периферических нервов
В случае локализации опухоли в доступном для пальпирования месте предположительный диагноз может быть определен неврологом после проведенного осмотра. Для его уточнения, а также в случае глубокого расположения новообразования необходимо УЗИ, МРТ мягких тканей. В случае опухоли на УЗИ обнаруживается округлое или веретенообразное образование, локализующееся внутри нервного ствола или тесно связанное с ним.
Невриномы характеризуются ровным контуром, пониженной эхогенностью, неоднородностью структуры; при длительном существовании могут содержать кисты и кальцификаты. Нейрофибромы отличаются более однородной структурой, могут иметь волнистый контур. МРТ позволяет более точно и детально визуализировать опухоль, определить ее границы.
С целью оценки степени нарушения проведения нервных импульсов по пораженному участку нерва осуществляется электронейрография. Пункционная биопсия опухоли периферических нервов не проводится, поскольку провоцирует ускоренный рост и малигнизацию новообразования. Гистологическое исследование возможно при взятии образца ткани опухоли во время операции по ее удалению.
Лечение опухолей периферических нервов
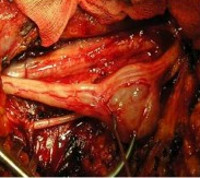
Основным способом лечения новообразований периферических нервных стволов является радикальное хирургическое удаление опухоли. Однако, учитывая частое рецидивирование таких образований и травматичность оперативного вмешательства, нейрохирург рекомендует операцию только при наличии показаний. К последним относятся интенсивный болевой синдром, выраженное нарушение проводимости по пораженному нерву, сдавление опухолью сосудистого пучка, приводящее к ишемии конечности. Решение об операции в случае нейрофиброматоза Реклингхаузена чаще отрицательное, поскольку удаление нейрофибромы зачастую приводит к ее рецидиву и провоцирует рост других имеющихся опухолей.
Оперативные вмешательства в отношении доброкачественных новообразований подразделяются на 3 метода: вылущивание опухоли, ее резекция вместе с участком нервного ствола, краевая резекция нерва с опухолью. Последние два метода проводятся с наложением шва нерва. В ряде случаев образование большого дефекта при иссечении участка нервного ствола делает невозможным выполнение эпиневрального шва и требует проведения пластики нерва. При признаках злокачественности образования (отсутствие границ между опухолью и пучками нервных волокон, невозможность определить оболочки нерва), подтвержденных результатами интраоперационной экспресс-биопсии, проводится иссечение нерва с опухолью до границы здоровых тканей. В запущенных случаях может потребоваться ампутация конечности.
Читайте также: