
Лейкоз хромосомных поломок нет
Острый лимфобластный лейкоз (ОЛЛ) — гетерогенная группа гемобластозов, развивающихся из самых молодых клеток лимфатического ряда. Эта группа состоит из отдельных подгрупп, каждая из которых в свою очередь гетерогенна.
Клоны клеток с хромосомными аномалиями обнаруживают примерно у 70—80 % пациентов, причем более чем в 30 % случаев это гипердиплоидные клоны, в 10 % — гиподиплоидные, в остальных — псевдодиплоидные.
Острый лимфобластный лейкоз (ОЛЛ) гораздо чаще наблюдается у детей и лиц молодого возраста, чем у пожилых. Средний возраст взрослых пациентов составляет около 30 лет. Мужчины болеют чаще, чем женщины.
Прогноз острого лимфобластного лейкоза у взрослых значительно хуже, чем у детей. Это коррелирует с возрастными различиями в частоте характерных аномалий кариотипа, т. е. при остром лимфобластном лейкозе у детей значительно чаще, чем у взрослых, встречаются хромосомные изменения, более благоприятные в прогностическом отношении, и наоборот — реже наблюдаются нарушения кариотипа, ассоциированные с плохим прогнозом. Особенно ярко возрастные различия проявляются при сравнении особенностей кариотипа острого лимфобластного лейкоза у детей и взрослых старше 60 лет.
Определенные особенности кариотипа важны для уточнения диагноза и прогнозирования ответа на терапию.
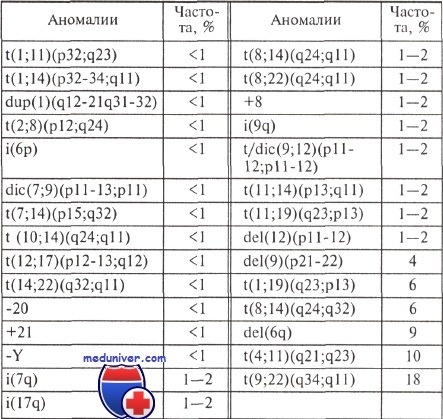
Хромосомные нарушения, наиболее характерные для острого лимфобластного лейкоза, представлены в таблице. Их частота вычислена S. Heim и F. Mitelman после объединения данных, полученных в разных цитогенетических лабораториях мира — всего около 6000 наблюдений. В таблице приведены не все, а только самые распространенные хромосомные аномалии, обнаруженные при остром лимфобластном лейкозе, но и они демонстрируют выраженную цитогенетическую гетерогенность этого заболевания. Большинство повторяющихся (неслучайных) аномалий наблюдается редко.
Необходимо отметить, что отдельные изменения кариотипа обнаруживают как при остром лимфобластном лейкозе, так и при других лимфоидных опухолях, другие изменения — исключительно при остром лимфобластном лейкозе. К первым относятся, в частности, перестройки хромосомных районов 14q11, 14q32, делеции длинного плеча хромосомы 6 и короткого плеча хромосом 9, 12 и 17. С другой стороны, t(9;22)(q34;q11), t(4;11)(q21;q23), t(1;19)(q23;p13) характерны для ОЛЛ, но не для других лимфопролиферативных заболеваний.
Еще одно общее замечание: В-клеточные лейкозы (в том числе и острые), а также лимфомы отличаются от Т-клеточных по спектру хромосомных изменений. Так, для новообразований В-клеточной природы характерны перестройки, затрагивающие локусы иммуноглобулиновых генов: гена Н-цепей иммуноглобулинов (14q32), L-цепей к (2р12) и h (2q11). В новообразованиях из Т-клеток нередко наблюдаются перестройки, затрагивающие гены Т-клеточных рецепторов (хромосомные участки 14q11, 7р15, 7q34).
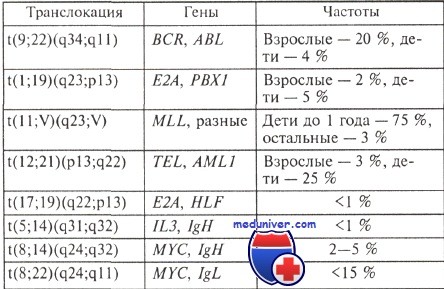
Частоты основных хромосомных перестроек, характерных именно для В- или Т-клеточных острых лимфобластных лейкозах, а также гены, вовлеченные в эти перестройки, приведены в таблицах.
Уже отмечалось, что частота отдельных характерных нарушений кариотипа различается при остром лимфобластном лейкозе у детей и взрослых. Это подтверждают данные таблице. Приведем конкретные примеры сначала по структурным изменениям, а затем по числовым.
Структурные аномалии: t(9;22) значительно чаще наблюдается у взрослых, a t(12;21) — у детей, причем первая крайне неблагоприятна в прогностическом отношении, а вторая — наоборот, предвещает хороший ответ на лечение в подавляющем большинстве случаев. Такие структурные аномалии, как перестройки хромосомного района 11q23, ассоциированные нередко с плохим прогнозом, часто наблюдаются у детей до 6 мес, их частота с возрастом понижается; у пожилых людей они редки.
Числовые изменения: клоны клеток с числом хромосом более 50 прогностически благоприятные, встречаются у детей значительно чаще, чем у взрослых: 25—30 и 5 % соответственно.
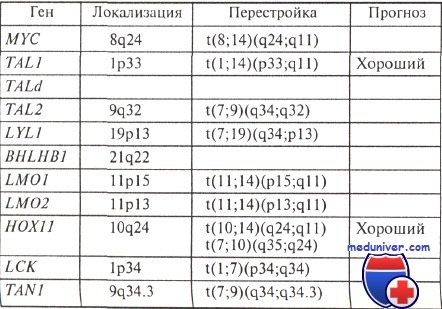
Числовые изменения кариотипа имеют большое значение для прогнозирования острого лимфобластного лейкоза. Уже отмечалось, что вариант острого лимфобластного лейкоза со значительным увеличением числа хромосом (более 50) имеет относительно благоприятный прогноз: 80—90 % больных переживает пятилетний срок с момента постановки диагноза, однако выявление некторых структурных перестроек в клетках лейкозного клона с такими числовыми нарушениями может ухудшить прогноз.
В отличие от острого лимфобластного лейкоза с гипердиплоидными клонами лейкозы, ассоциированные с гиподиплоией, имеют обычно плохой прогноз: только 30 % больных остается в живых по истечении 2 лет от момента постановки диагноза. Еще более неблагоприятен прогноз при острых лейкозах с так называемым окологаплоидным числом хромосом, при которых каждая клетка содержит 26—28 хромосом. В таких клетках вместо большинства хромосомных пар остается по одному гомологу; сохранено по два гомолога только в 10-й, 14-й, 18-й и 21-й парах.
Неблагоприятен прогноз и в случаях с околотетраплоидными клонами. Следует отметить, что окологаплоидный и околотетраплоидные клоны клеток редко наблюдаются при остром лимфобластном лейкозе — не более 1—2 % случаев.
Остановимся на отдельных структурных аномалиях кариотипа, характерных для острого лимфобластного лейкоза.
Филадельфийская хромосома характерна для ХМЛ или хронического миелоидного лейкоза, который является клональным новообразованием, развивающимся из кроветворных стволовых клеток.
Данная опухоль была первой, при которой обнаружили признаки характерного хромосомного маркера. Открытие в 1960 году в Филадельфии сделали американские исследователи D. A. Hungerford и P. C. Nowell, в связи в чем этот маркер назвали термином филадельфийская хромосома-Ph. И именно эта находка послужила началом клинической цитогенетики в онкологии.
Что собой представляет хромосома?
Может появляться и при других видах лейкоза. Филадельфийская хромосома является укороченной хромосомой, входящей в группу малых акроцентриков. Каждая нормальная женская клетка этой группы содержит их 2 пары– 21 и 22, клетка же мужская включает не четыре, а пять таких хромосом, потому что помимо 21 и 22 пар включается еще и Y-хромосома.
Без G-бендинга, то есть при обычном окрасе, Ph-хромосома выявляется почти у каждого пациента, страдающего ХМЛ, а именно в 95-98 процентах случаев. На хромосомах, которые окрашены дифференциально, видно, что одна из 22 пар является укороченной.
Каков процент обнаружения?
Приблизительно у 90 % пациентов ее видно в каждой анализируемой метафазе, а у остальных больных обнаруживаются и клетки с хромосомой-Ph, и клетки нормальные.
К тому же в некоторых случаях Ph-хромосому регистрируют в меньшинстве клеток костного мозга. Транслокацию (9;22) наблюдают в мегакариоцитах, миелоидных клетках, В- и Т-лимфоцитах, эритробластах. Данный факт является свидетельством того, что болезнь берет начало с предшественницы гемопоэза – некоммитарованной клетки.
Атипичные транслокации
Приблизительно в 10 % случаев можно наблюдать атипичные транслокации, когда цитологическое стандартное исследование может позволить увидеть перенос части 22 хромосомы не на 9, а на любую другую. Более того, в некоторых случаях при ХМЛ обнаруживаются сложные Ph-транслокации при участии не 2 (22 и 9 хромосом), а 3 хромосом или большего количества.

Установили, что почти при каждой Ph-транслокации участвуют 9 и 22 хромосомы, но не всегда это можно увидеть при проведении стандартного цитогенетического исследования, однако это обнаруживают во время использования ПЦР и FISH.
Многочисленные исследователи полагают, что тип Ph-транслокации (сложная, атипичная, стандартная) клинического значения не имеет.
Разрыв генов
Использование методов молекулярно-генетических поспособствовало установлению того, что разрыв в 9-й хромосоме проходит через протоонкоген (ген ABL), который раньше был идентифицирован у мышей в одном из вирусов лейкоза. В 22-й хромосоме наблюдают разрыв BCR гена. Результатом слияния фрагментов генов BCR и ABL становится образование химерного гена BCR-ABL расположенного обычно на 22-й делетированной хромосоме.
Как вызывает филадельфийская хромосома лейкоз?
Приблизительно 70 % случаев характеризуется обнаружением помимо транскрипта BCR-ABL продукта другого химерного гена, который образуется вследствие t (9;22) —ABL-BCR на der (9), но роль его для развития хронического миелолейкоза не выяснена.
Делеция
Также установили очень важный факт: приблизительно 20-25 % пациентов, у которых обнаружен хронический миелолейкоз, имеют делецию в маркерной 9q+ хромосоме. Эту аномалию нельзя обнаружить при проведении простого хромосомного анализа, но можно увидеть ее во время использования FISH с зондами, которые специально разработаны.
Размер делеции варьируется у каждого пациента: участок с делецией может содержать последовательности BCR-гена, которые перенесены с 22-й хромосомы на 9-ю, на последовательности самой 9 хромосомы или обеих.
Установили также возникновение делеции с формированием специфической t (9;22) одновременно, что частота ее в группах пациентов, которых обследовали на различных стадиях хронического миелолейкоза, одинаковая.
Пока что применение данного метода диагностики (информативного прогностического), к сожалению, в широкую клиническую практику не вошло в связи с его сложностью и требованием дорогостоящего оборудования и реактивов.
Ее роль в прогрессии ХМЛ
Делеция маркера 9q+ играет важную роль в прогрессии ХМЛ, но до конца все еще не выяснена. Когда выпадают кодирующие последовательности генов ABL или/и BCR вследствие делеции, это приводит к экспрессированию лишь одного химерного гена BCR-ABL, но экспрессии гена ABL-BCR не происходит. Вероятно, данное событие также играет важную роль в прогрессировании лейкоза. Более того, исследователи обсуждают возможность инактивации пока еще неизвестных генов-супрессоров, которые локализованы в хромосомном делетируемом районе.
Белок с мол. м. 210 тысяч кодируется химерным геном BCR-ABL, который обладает большей, нежели продукт протоонкогена нормального ABL (H145), протеинкиназной активностью. Во время лейкоза, который у мышей вызван вирусом Абельсона, онкогенную активность имеет белок, продукт gag/abl гибридного гена, который имеет высокую протеинкиназную активность. В эксперименте проводилось вырезание gag/abl гена, после чего вирус уже не мог у мышей вызывать лейкоз. То есть филадельфийская хромосома является маркером этого заболевания.
Подробнее о генах BCR и ABL
При изучении в генах BCR и ABL разрывов при ХМЛ было выявлено, что у разных пациентов разная их локализация. К примеру, в ABL гене протяженность участка, где могут быть разрывы, большая, до 200 kb, а в BCR-гене локализация разрывов происходит на небольшом участке в 8,5kb, то есть можно говорить о наличии кластера разрывов, давшего название самому BCR гену - Breakpoint cluster region.
Во многих случаях t (9;22) разрывы BCR-гена обнаруживаются на участке M-BCR, причем в химерный ген включается длинная часть BCR гена, результатом чего становится появление белка, характерного для ХМЛ P210Bcr/Abl. К тому же при t (9;22) разрывы BCR-гена локализоваться могут на участках, которые называются m-bcr и u-bcr. Bm-bcr области разрывы ведут к образованию P190Bcr/Abl химерного белка, то есть меньшего по величине, чем Р210Bcr/Abl. А локализация разрывов в u-bcr ведет к образованию белка Р230Bcr/Abl более крупного.
Эти различия молекулярного характера коррелируют не строго с клинической особенностью лейкоза. Например, Р190Bcr/Abl имеется у 2 разных видов лейкозов: Ph-позитивный лимфобластный острый лейкоз и гранулоцитарный хронический лейкоз при выраженном моноцитозе и миелопластическими чертами. Если обнаруживают Р230Bcr/Abl, то можно наблюдать картину нейтрофильного хронического миелолейкоза, то есть миелолейкоз, формула крови при котором представлена единичными метамиелоцитами и зрелыми нейтрофилами. К тому же название нейтрофильного гранулоцитарного лейкоза долгое время используют, чтобы обозначать Ph(BCR-ABL)-негативный хронический миелолейкоз (относительно доброкачественный вариант, наблюдаемый у пожилых людей и иногда у подростков).
Любой из вышеприведенных белков можно обнаружить при хроническом миелолейкозе. Также сообщается о достаточно частом сочетании 2 типов белков (Р1900Bcr/Abl и Р210Bcr/Abl) при лимфобластном остром лейкозе и хроническом типичном миелолейкозе.
Роль BCR-ABL для развития хронического миелолейкоза
Решающую роль BCR-ABL гена и его продукта, а именно Р210 белка для развития хронического миелолейкоза демонстрировали на разных системах in vitro и in vivo. Например, при трансдукции bcr/abl в гемопоэтические стволовые клетки мышей и их дальнейшей трансплантации сингенным облученным животным у вторых возникает миелопролиферативная болезнь, похожая на хронический миелолейкоз человека. Установили, что онкогенный потенциал обуславливает высокая тирозинкиназная активность химерного белка BCR-ABL. Одним из центральных в злокачественной трансформации клеток является событие дерегуляции тирозинкиназной активности.
Когда исследователи вводили летально облученным мышам клетки, которые экспрессировали р210Bcr/Abl, то одни животные заболевали лейкозом, сходным с человеческим хроническим миелолейкозом, а другие различными новообразованиями из клеток гемопоэтических: эритроидные опухоли, миеломоноцитарные лейкозы, ретикулоклеточные саркомы, пре В- и Т-клеточные лимфомы, макрофагальные опухоли. Причину различий не выяснили. Такие опыты показывают, что всю цепь событий, приводящую к развитию хронического миелолейкоза еще не установили. Но факт остается. При этом заболевании обнаруживается филадельфийская хромосома в клетках костного мозга. Лечение рассмотрим ниже.
Что еще обнаружили у больных?
Получили данные, которые свидетельствуют о существенном увеличении массы кроветворных клеток и элементов крови в организме пациентов с хроническим миелолейкозом, в основном из-за резкого увеличения срока жизни таких клеток, потому что активированным геном ABL (в BCR-ABL гене) ингибируется апоптоз – запрограммированная клеточная смерть. К тому же этим геном усиливается пролиферация миелоидных клеток.
Имеются основания полагать, что в случае хронического миелолейкоза изменяется функция спец. клеточных белков, то есть интергинов, последствием чего становится нарушение адгезии к стромальным элементам молодых миелоидных клеток, а также происходит избегание стволовых лейкемических клеток негативных регуляторных влияний. Можно сделать вывод. Наличие филадельфийской хромосомы патогномонично для хронического миелолейкоза.
Выводы

Наиболее общей формулировкой молекулярного патогенеза является следующая: химерным геном BCR-ABL кодируется белок, у которого постоянно активирована тирозинкиназная активность, что приводит к активации большого количества сигнальных путей, а также выраженным изменениям апоптоза, адгезии и клеточного цикла. Считается, что эти события достаточны для определения злокачественной клеточной трансформации и поддерживания опухолевого фенотипа.
Но все равно основным маркером является филадельфийская хромосома.
Лечение
Для подавления активности патологических лейкоцитов необходимой для сохранения жизни больных им требуется вторая линия терапии. Для этого применяют нилотиниб – вещество, которое блокирует передачу сигнала от филадельфийской хромосомы.
Ведь именно это и заставляет костный мозг продуцировать в большом количестве поврежденные лейкоциты.
Специалистами выделяется 3 ступени. Во время первой достигается гематологическая ремиссия, нормализация анализа крови и состояния пациента (размера его селезенки).
Но в клетках все же остается филадельфийская хромосома. Во вторую очередь достигается цитогенетическая ремиссия, когда эта хромосома не определяется в клетках. В-третьих, при проведении молекулярного исследования клеток крови в результате воздействия ингибиторами тироксинкиназы у костного мозга не обнаруживают патологический ген.
Трансплантация костного мозга
Обязательна ли ТКМ при филадельфийской хромосоме? Такую операцию по трансплантации проводят больным с острой формой миелоидного лейкоза. Также реально совместимого донора найти бывает непросто. Это очень серьезная и длительная операция. Но она позволяет восстановить нормальную работу костного мозга.
Изучение хромосомных аномалий при остром лпмфобластном лейкозе (ОЛЛ) представляется крайне важной задачей. Цитогенетический анализ случаев ОЛЛ долгое время был связан со значительными методическими трудностями из-за плохой морфологии метафазных хромосом, патогномичной для лимфобластных клопов. Усовершенствование методик позволило установить изменения кариотипа примерно у 70% (55-90%) больных, причем, в отличие от других форм лейкозов, модальное число хромосом при ОЛЛ — самостоятельный прогностический признак.
У 25-30% пациентов клоны клеток содержат более 50 хромосом. Этот кариотип ассоциирован с рге-В/соттоп иммунофенотипом.
Гипердиплоидия (и > 50 хромосом), как правило, ассоциируется с наибольшей продолжительностью первой ремиссии и высокой безрецндивной выживаемостью (медиана составляет 50 месяцев). Предполагается, что гипердиплоидные клетки высокочувствительны к терапии фазовоспецифичными противоопухолевыми препаратами вследствие большой продолжительности 8-фазы. Почти в половине случаев гипердиплоидии п > 50 обнаруживаются дополнительные структурные хромосомные аберрации. Чаще других встречаются: !(9;22), !(4; 11) и 18о(17д). Наличие в кариотипе опухолевых клеток перечисленных неслучайных структурных ано-
малий обусловливает у таких пациентов возникновение ранних рецидивов или плохой ответ на терапию.
Промежуточный прогноз заболевания характерен для группы больных с модальным числом хромосом в лейкозных клетках — от 47 до 50 (гипердиплоидия п = 47-50). Довольно сложно оценить прогноз у пациентов с околотриплоидным набором хромосом из-за недостаточного числа подобных наблюдений. По некоторым данным, случаи с околотетраплоидным набором хромосом часто ассоциируются с Т-клеточным фенотипом ОЛЛ и могут иметь неблагоприятный прогноз. Наиболее часто в трисомии вовлекаются следующие хромосомы: 4, 6, 10, 14, 17, 18, 21 и X. Дополнительные структурные аномалии, как правило, являются типичными для ОЛЛ.
иммунологически фенотипом лейкоза. Ьашрей с соавторами обнаружили у больных с 1(4;1 1) перестройку генов тяжелых цепей иммуноглобулинов, что позволяет говорить о В-клеточной природе заболевания. Описаны также врожденные ОЛЛ с 1(4;11). При наличии у пациентов этой аберрации прогноз заболевания крайне тяжелый. Ремиссии удается достичь примерно у 50-60% больных, а ее продолжительность составляет всего 3 месяца. Для мониторинга минимальной резидуальной болезни можно использовать методы ПЦР и Р18И.
Для больных ОЛЛ характерны нарушения кариотипа, затрагивающие хромосому 8-й пары, где расположен с-тус онкоген, и области локализации генов тяжелых и легких цепей иммуноглобулинов - 2р12,14д32 и 22д11. Образуется !(2;8)(р12;д24) и ее варианты: !(8;14)(д24;д32), !(8;22)(д24;д11), которые приводят к активации онкогена с-тус, увеличивающейся его транскрипции и, в конечном счете, к неопластической трансформации. Иммунологически характерен В-клеточный вариант. Для пациентов с описанными выше аберрациями типичны поражение ЦНС, гепатоспле- номегалия, лимфаденопатия, резко повышенные уровни мочевой кислоты и лактатдегидрогеназы в крови. Ранее такие нарушения кариотипа относили к прогностически неблагоприятным хромосомным аномалиям. Используя современные протоколы терапии, полную ремиссию удается достичь у 86% взрослых больных, а
3- летняя выживаемость у них составляет 74%.
Для больных с Т-клеточным вариантом ОЛЛ типичными являются изменения кариотипа, затрагивающие точки локализации ге- ПНр://шшш.Ье51тедЬоок.сот/
| 1 1 ! ' '8 * 1 'I в М1 й 1 •* «1 :'Д л
'4 А 1 '* в 1 'А 0 1 'ХА' >Д 4. 1
19 20 21 22 г у
Рис. 4. Кариограмма пациента С. Ю. В.: 46, ХУ. де18(д24)
нов Т-клеточных рецепторов. Примером может служить 1(8; 14) (д24;д11), при которой с-тус онкоген патологически экспрессирован. В результате слияния генов МУС, картированного на 8(]24, и ТСКО, локализованного на 14д, образуется химерный ген, кодирующий протеин с новыми свойствами. Эта транслокация обусловливает крайне тяжелый прогноз заболевания (медиана выживаемости составляет 5 месяцев).
Приблизительно у 5% больных ОЛЛ встречается изолированная делеция Йе18(д24), которая также ассоциируется с крайне неблагоприятным прогнозом заболевания (рис. 4).
Делеция длинного плеча 6 хромосомы (йе1 6(д21)) (рис. 5) является характерным нарушением для ОЛЛ и встречается у 5-10% пациентов. На уровне 6д21-24 располагается онкоген с-шуЬ, гиперэкспрессия которого обнаруживается во всех случаях заболеваний с Йе16(д21). Большей частью аномалия встречается при соттоп ОЛЛ. Прогноз заболевания при данном нарушении кариотипа относительно благоприятный, медиана выживаемости составляет около 38 месяцев.
Однако описываются случаи обнаружения йе16(д) и при ОНЛЛ и ХМЛ. В этих случаях пациенты часто имеют дополнительные хромосомные нарушения. -
Данная аномалия обнаруживается при всех вариантах ОНЛЛ, но чаще наблюдается при ОНЛЛ Мб (26% всех ед-ОНЛЛ случаев). Как правило, уровни поломок находятся на д12 27. причем в 80% наблюдений вовлекается 6д21—23 район. Экспрессия с-туЬ обнаруживается во всех случаях.
| Рис. 6. Кариограмма пациентки Я.Р. А.: 46, XX, 1(1; 19)^23;р13) |
Транслокация 1(1;19)(я23;р13) (рис. 6) встречается в 3% всех случаев ОЛЛ с кариологическими нарушениями.
Это нарушение ассоциируется с рге-Б-ОЛЛ фенотипом. Молекулярные особенности 1(1; 19) в настоящее время до конца не изучены. Однако известно, что аберрация приводит к слиянию Е2А
гена, картированного на 19р13, с РВХ1 геном, расположенным на 1д23. Беёега и соавторы обнаружили в экспериментах на животных, что данный химерный ген индуцирует не только пролиферацию и апоптоз, но и злокачественные лимфомы у животных.
Прогностические особенности 1(1; 19) не совсем ясны, однако большинство исследователей показывают, что данная аберрация обусловливает неблагоприятный исход заболевания. У большинства больных рецидив наступает в течение года после достижения ремиссии.
Транслокация !(12;21)(р13;д22) — наиболее часто встречающаяся при В-клеточном ОЛЛ аномалия кариотипа у детей. Она является криптической (скрытой), не выявляющейся при стандартном цитогенетическом исследовании. Данная аберрация выявляется при помощи молекулярно-генетических методов исследования в 19-27% детских ОЛЛ. У взрослых описаны лишь единичные случаи. На молекулярном уровне вследствие транслокации образуется химерный ген из генов ТЕЬ (12р13) и АМЫ (2Ц22). Клинически данная транслокация ассоциируется с возрастом больных от
1 до 10 лет, относительно невысоким лейкоцитозом (
ОСТРЫЕ ЛЕЙКОЗЫ
Классификация. Этиология. Патогенез.
А.В.Колосков
(лекция для врачей и студентов)
Под термином "острые лейкозы" понимают группу клональных заболеваний, первично возникающих в костном мозге в результате мутации стволовой клетки крови. Следствием мутации является потеря потомками мутировавшей клетки способности к дифференцировке до зрелых клеток крови.
Группу острых лейкозов объединяет общий морфологический признак: субстрат опухоли представлен незрелыми молодыми клетками -- бластами. Классификация острых лейкозов основана на признаках принадлежности опухолевых клеток к тому или иному ростку гемопоэза. Принадлежность опухолевых клеток может быть определена цитохимическим методом на основании выявления в цитоплазме этих клеток специфических включений (например гликогена в клетках лимфоидного ростка гемопоэза, миелопероксидазы в клетках миелоидного ростка гемопоэза или альфа-нафтилэстеразы в клетках моноцитарного ряда). Кроме того, для определения гистогенеза опухолевых клеток используется иммунологический метод (иммунофенотипирование), выявляющий на цитоплазматической мембране клетки антигены (кластеры дифференцировки -- CD), указывающие на происхождение клетки и степень ее зрелости.
На сегодняшний день для практических и научно-исследовательских целей используется Франко-Америко-Британская (FAB) классификация острых лейкозов. Классификация подразделяет все острые лейкозы на две главные подгруппы -- острые нелимфобластные лейкозы (составляют около 70% всех острых лейкозов) и острые лимфобластные лейкозы (составляют 30% всех острых лейкозов).
Для разграничения различных вариантов острых лейкозов (ОЛ) FAB классификация использует ряд цитологических критериев аспирата костного мозга и мазка периферической крови, а также цитохимические тесты.
Первым шагом FAB классификации является разграничение ОЛ и миелодиспластического синдрома, а также выделение острого эритробластного лейкоза (для его обозначения FAB классификация использует символ -- М6) (рисунок 1).
Рисунок 1. Алгоритм для диагностики острых лейкозов и разграничения их с миелодиспластическим синдромом. (по Bennett J.M. et al., 1985).
Далее для на основании цитологических и цитохимических критериев FAB классификация выделяет следующие варианты ОНеЛЛ (по Bennett J.M. et al., 1985).
Острый миелобластный лейкоз (М1)
Аспират костного мозга:
- бластные клетки составляют не менее 90%;
- созревающие гранулоциты (под этим термином FAB-классификация понимает все гранулоцитарные клетки от промиелоцитов до сегментоядерных) составляют менее 10%.
Острый миелобластный лейкоз с частичным созреванием (М2)
Аспират костного мозга:
- бластные клетки составляют не менее 30%, но менее 90%;
- клетки моноцитарного ростка кроветворения составляют менее 20%;
- созревающие гранулоциты составляют не менее 10%;
Острый промиелоцитарный лейкоз (М3)
Этот вариант ОНеЛЛ устанавливают на основании типичного морфологического субстрата (характерного вида промиелоциты в аспирате костного мозга) без использования каких-либо дополнительных тестов.
Острый миеломонобластный лейкоз (М4)
1. Аспират костного мозга:
- клетки миелоидного ростка кроветворения составляют не менее 30%, но менее 80%.
2. Периферическая кровь:
- клетки моноцитарного ростка кроветворения составляют не менее 5.0 х 10^9/л;
- Если 1 и 2 условия выполнены, то устанавливают диагноз -- М4 вариант ОНеЛЛ.
- Если 1 условие выполнено, а 2 не выполнено, то оценивают результаты цитохимической окраски бластных клеток на альфа-нафтилэстеразу. Если определяется не менее 20% бластов, дающих положительную окраску на альфа-нафтилэстеразу, то устанавливают диагноз -- М4 вариант ОНеЛЛ.
- Если определяется менее 20% бластов, дающих положительную окраску на альфа-нафтилэстеразу, то устанавливают диагноз -- М2 вариант ОНеЛЛ.
- Если аспират костного мозга соответствует описанию при М2 варианте ОНеЛЛ, а 2 условие выполнено, то оценивают результаты цитохимической окраски бластных клеток на альфа-нафтилэстеразу.
- Если определяется не менее 20% бластов, дающих положительную окраску на альфа-нафтилэстеразу, то устанавливают диагноз -- М4 вариант ОНеЛЛ.
- Если определяется менее 20% бластов, дающих положительную реакцию на альфа-нафтилэстеразу, то устанавливают диагноз -- М2 вариант ОНеЛЛ.
- Если в аспирате костного мозга присутствует не менее 5% эозинофилов, то устанавливают диагноз М4э вариант ОНеЛЛ (острый миеломонобластный лейкоз с эозинофилией).
Острый монобластный лейкоз (М5)
Аспират костного мозга:
-- клетки моноцитарного ряда гемопоэза составляют не менее 80%.
- Если монобласты составляют более 80% от всех клеток моноцитарного ростка кроветворения, то устанавливают диагноз -- М5а вариант ОНеЛЛ (острый монобластный лейкоз без созревания).
- Если монобласты составляют менее 80% от всех клеток моноцитарного ростка кроветворения, то устанавливают диагноз -- М5b вариант ОНеЛЛ (острый монобластный лейкоз с созреванием).
- В дополнениях к рассмотренной редакции FAB классификации выделяют еще два варианта ОНеЛЛ.
Острый мегакариобластный лейкоз (М7) -- диагноз устанавливают на основании данных электронно-микроскопического исследования бластных клеток или на основании данных иммунофенотипирования.
Острый ранний миелобластный лейкоз (М0) - диагноз устанавливают на основании отрицательных результатов всех цитохимических окрасок бластных клеток или на основании данных иммунофенотипирования.
Для классификации ОЛЛ FAB классификация использует цитологические особенности бластных клеток. На основании этих признаков проводится подразделение на три варианта - L1, L2 и L3. Однако такое классификационное построение оказалось условным. Выделенные варианты реально не отличались по особенностям течения, длительности выживания больных и ответу на терапии, что послужило основанием для отказа от этой части FAB классификации. В настоящее время используются иммунофенотипическая классификация ОЛЛ, которая выделяет три основное группы:
- Т-клеточный острый лимфобластный лейкоз (опухолевые клетки несут на своей поверхности антигенные маркеры принадлежности к Т-ряду лимфопоэза);
- В-клеточный острый лимфобластный лейкоз (опухолевые клетки несут на своей поверхности антигенные маркеры принадлежности к В-ряду лимфопоэза);
- общий острый лимфобластный лейкоз (опухолевые клетки при этом варианте лейкоза несут на своей поверхности антиген, специфичный для лимфоидных предшественников -- общий антиген острого лимфобластного лейкоза).
Эпидемиология.
Ежегодно регистрируется 35 новых случаев острых лейкозов на 1 млн населения. Структура встречаемости острых лейкозов в значительной степени зависит от возраста. Так в возрастной группе до 15 лет соотношение ОЛЛ : ОНеЛЛ составляет 4 : 1, в возрастной группе от 15 до 35 лет - 1 : 1.5, а в возрастной группе старше 35 лет - 1 : 8. Мужчины и женщины болеют с одинаковой частотой.
Для острых лейкозов, как и для большинства других опухолевых заболеваний, невозможно выделить специфический этиологический фактор. Этиологические факторы, способные вызывать развитие опухоли, были подробно рассмотрены в главе "Хронические миелопролиферативные заболевания".
В основе патогенеза острых лейкозов лежит мутация стволовой клетки крови, что влечет за собой практически полную потерю потомками мутировавшей клетки способности к созреванию. Мутантный клон автономен от каких-либо регулирующих воздействий организма и достаточно быстро вытесняет нормальные гемопоэтические клетки, замещая собой весь гемопоэз.
С момента мутации до момента появления клинических и лабораторных признаков заболевания проходит в среднем 2 месяца. За этот период времени количество опухолевых клеток увеличивается с 1 (родоначальница мутантного клона) до 10^9 -- 10^12. Масса такого количества клеток составляет около одного килограмма. Вытеснение нормальных гемопоэтических клеток, и замещение их опухолевыми клетками, неспособными к созреванию, закономерно приводит к уменьшению в периферической крови зрелых клеток с развитием анемии, гранулоцитопении, лимфопении, моноцитопении и тромбоцитопении, что будет проявляться соответствующей клинической картиной.
Уменьшение количества эритроцитов влечет за собой развитие анемического синдрома. Уменьшение или полное исчезновение зрелых гранулоцитов, приводит к развитию иммунодефицита и инфекционных осложнений. Свой вклад в патогенез инфекционных осложнений вносят также лимфопения и моноцитопения. Тромбоцитопения лежит в основе кровоизлияний и кровотечений.
В ряде случаев опухолевые клетки не нуждаются в строго необходимом для нормальных гемопоэтических клеток стромальном микроокружении. Они могут покидать костный мозг и образовывать колонии опухолевого гемопоэза в других органах и тканях (селезенке, лимфоузлах, печени, центральной нервной системе, легких, коже, слизистых оболочках). Инфильтрация опухолевыми клетками органов и тканей относят к проявлениям пролиферативного синдрома. Степень злокачественности опухолевых клеток при остром лейкозе с течением времени возрастает (как и для других групп опухолей, для острых лейкозов правомочен закон опухолевой прогрессии). Поскольку опухолевые клетки при острых лейкозах, в большинстве вариантов, изначально имеют выраженный дефект созревания, то большая злокачественность часто проявляется появлением экстрамедуллярных очагов кроветворения, увеличением пролиферативной активности, развитием резистентности к проводимой терапии. В основе озлокачествления лежат вторичные мутации в опухолевых клетках. Это наглядно демонстрируют результаты цитогенетического исследования, выявляющего на ряду с изменениями хромосом, имевшими место в начале заболевания, появление дополнительных поломок по мере течения заболевания.
В дебюте острых лейкозов хромосомные аномалии выявляют в 90% случаев. Однако, столь же часто выявляемой хромосомной поломки, как транслокация 9;22 (филадельфийская хромосома) при хроническом миелолейкозе, при острых лейкозах не наблюдается. Тем не менее известна связь некоторых перестроек хромосом с вариантом острого лейкоза. Так транслокация 15;17 является специфичной для острого промиелоцитарного лейкоза (М3), она выявляется в среднем в 50% случаев. Транслокация 8;21 чаще всего встречается при остром миелобластном лейкозе (М1). В трети случаев общего варианта ОЛЛ встречается транслокация 9;22 (филадельфийская хромосома). Клоны клеток с аномалией хромосом не определяются в период ремиссии и появляются вновь при развитии рецидива заболевания. Наиболее часто встречающиеся при острых лейкозах поломки хромосом представлены в таблице 1.
Таблица 1. Наиболее частые аномалии хромосом при острых лейкозах.
хромосомная аномалия вариант острого лейкоза
| хромосомная аномалия | вариант острого лейкоза |
| t(8;21) | M2 |
| t(15;17) | M3 |
| inv(16) | M1;M2 |
| t(9;22) | M1;M2 |
| t(6:9) | M2;M4 |
| t(9:11) | M4;M5 |
| t(8;16) | M5b |
| inv(3) | M1;M2;M4;M7 |
| -7/7q- | M1;M2;M3:M4:M5 |
| 5q- | M1;M2;M3;M4; |
| t(3;5) | M2;M6 |
| t(9;22) | общий вариант ОЛЛ |
| t(7;12) | В-варианты ОЛЛ |
| t(9;12) | -"- |
| t(1;19) | -"- |
| t(8;14) | -"- |
| t(8;12) | -"- |
| t(2;8) | -"- |
| t(11;14) | Т-варианты ОЛЛ |
| t(10;14) | -"- |
| t(8;14) | -"- |
| t(1;14) | -"- |
| inv(14) | -"- |
| 7 | -"- |
| 7q+ | -"- |
Таким образом, в основе патогенеза острых лейкозов лежит мутация стволовой клетки крови. Следствием мутации является развитие в костном мозге клона клеток, утративших способность к созреванию. Неопластический клон вытесняет нормальные гемопоэтические клетки, что приводит к развитию дефицита зрелых клеток в периферической крови. Снижение количества или полное отсутствие зрелых клеток периферической крови приводит к выпадению соответствующих функций периферической крови, что влечет за собой развитие клинических проявлений заболевания. Патоморфология костного мозга.
В гистологическом препарате костного мозга при острых лейкозах определяется значительное увеличение количества бластных клеток, которые часто заполняют весь препарат .
Читайте также: