
Хроническое миелопролиферативное заболевание истинная полицитемия

В норме в губчатом веществе костного мозга, находящегося в полостях крупных костей скелета, продуцируются незрелые стволовые клетки. Постепенно они созревают и преобразуются в полноценные форменные элементы:
- эритроциты, доставляющие кислород к органам и тканям,
- лейкоциты, защищающие организм от инфекционных агентов и прочих чужеродных веществ,
- тромбоциты, формирующие кровяной сгусток и останавливающие кровотечение.

созревание форменных элементов крови в норме
При наличии у человека миелопролиферативного заболевания в кровь поступают клетки, неспособные выполнять свои функции. Стволовые клетки при патологии часто преобразуются только в один тип форменных элементов. Патологический процесс отличается медленным прогрессированием.
Миелопролиферативное заболевание — это собирательное понятие, включающее группу гемобластозов, которые характеризуются аномальным разрастание костномозговых структур, отвечающих за образование кровяных телец. Выделяют несколько основных форм МПЗ, при которых поражаются разные клеточные элементы крови:
- Истинная полицитемия,
- Эссенциальная тромбоцитемия,
- Хронический миелолейкоз.
Причины

пример jak2 мутации при истинной полицитемии (избытке эритроцитов)
В основе МПЗ лежит приобретенная генная мутация, обусловленная влиянием негативных внешних или внутренних факторов. Мутация генов MPL и jak 2 приводит к повреждению ДНК одной гемопоэтической клетки, которая дает начало всем типам клеточных элементов. Аномально изменившаяся бластная форма приобретает отрицательные черты — перестает развиваться, не созревает полностью, не самоуничтожается, а непрерывно делится и порождает многочисленные клоны. Именно поэтому МПЗ называют клональным. Клоны также остаются на начальном уровне развития и имеют полностью недифференцированную структуру. Повреждаться может как один, так сразу несколько ростков кроветворения.
В результате в костном мозге увеличивается количество клеток-предшественниц эритроцитарного, тромбоцитарного и лейкоцитарного типов. По мере их накопления в кровяном русле ухудшается самочувствие больных. От того, какой росток переродился, зависит характер патологии, ее симптоматика и прогноз. Формы МПЗ отличаются медленным развитием. Если заболевание было выявлено на ранней стадии, у больного есть все шансы добиться стойкой ремиссии.
Причины, вызвавшие мутационные процессы, остаются до конца неизученными. Одни ученые относят к ним негативные факторы окружающей среды, другие — ошибки при делении клеток. МПЗ не является наследственным. Мутации генов могут возникать на протяжении всей жизни человека. Они называются приобретенными. Риск развития патологии увеличивается с возрастом. Лицам старше 50 лет необходимо внимательно относиться к здоровью и при появлении подозрительной симптоматики обращаться к гематологу. Вероятность развития недуга повышается под воздействием факторов риска — облучения и химикатов, оказывающих токсическое влияние на организм.
Классификация
Миелопролиферативные заболевания имеют код по МКБ 10 — D47.1. По типу течения их подразделяют на острые и хронические. В первую группу входят максимально агрессивные и быстро прогрессирующие недуги, поражающие в основном молодых людей. К группе хронических миелопролиферативных заболеваний относятся медленно развивающиеся патологии, имеющие относительно благоприятный прогноз и возникающие у пожилых лиц.
В зависимости от пораженного ростка кроветворения выделяют следующие формы процесса:
- Истинная полицитемия — гиперпродукция красных кровяных телец и сгущение крови. Эритроциты задерживаются в селезенке, развивается спленомегалия. У больных возникают признаки тромбогеморрагического синдрома, повышается риск инсультов и инфарктов. В целом данная форма отличается доброкачественным течением. По сравнению с другими видами МПЗ она характеризуется высокой выживаемостью.
- Эссенциальный тромбоцитоз — опасное для жизни состояние, при котором происходит усиленное образование тромбоцитарных клеток.
- Хронический миелолейкоз — злокачественное заболевание, характеризующееся преимущественным поражением гранулоцитарного ростка и появлением в крови недифференцированных лейкоцитов.
- Эозинофильная лейкемия — усиленное разрастание и повреждение эозинофилов, которые относятся к лейкоцитарным клеткам. При этом нарушаются их главные функции — борьба с инфекцией и иммунный ответ на потенциальные аллергены.
- Миелофиброз — образование в косном мозге патологически измененных клеток с заменой функциональной ткани соединительнотканными волокнами.
- Хронический нейтрофильный лейкоз — формирование незрелых нейтрофилов, которые перестают защищать организм от патогенов.

Классификация МПЗ имеет важное значение для диагностики онкологических заболеваний органов кроветворения. С ее помощью гематологи-онкологи могут легко определить тип сформировавшейся патологии и подобрать больному адекватную терапию, которая может спасти жизнь.
Видео: лекция по классификации и патогенезу ХМПЗ
Развитие и симптомы
Существует три пути распространения заболевания по организму:
- Лимфогенный — аномальные структуры проникают во внутренние органы по лимфатическим сосудам.
- Гематогенный — проникновение видоизмененных клеток в здоровые ткани по кровеносному руслу.
- Имплантационный — прорастание пораженных бластных форм в соседние органы и близлежащие ткани.
Гематогенное распространение злокачественных клеток считается самым опасным. Таким пациентам вместе с лечебными мероприятиями проводят динамическое наблюдение за функционированием внутренних органов. Данный тип патологии дает метастазы в самые отдаленные участки организма человека, что приводит к формированию вторичных онкологических очагов.
Клиническая картина МПЗ зависит от конкретной формы процесса, сопровождающегося разрастание кроветворных тканей костного мозга и чрезмерным поступлением в кровоток остановившихся в своем развитии атипичных кровяных телец. Каждый вид заболевания отличается характерной симптоматикой. Но существуют общие распространенные симптомы. Это признаки анемии или тромбоза:
- не проходящая слабость, быстрая утомляемость, упадок сил,
- отсутствие аппетита и похудание,
- шум в ушах и головокружение,
- помрачение сознания,
- дезориентация во времени и пространстве,
- гематомы на теле,
- частые кровотечения и кровоизлияния,
- отечность тканей и артралгия,
- абдоминальная боль,
- бледность кожи,
- гепатоспленомегалия,
- плетора (“полнокровие”),
- лихорадка.
Это общая симптоматика, возникающая при любой форме МПЗ. Существуют также специфические проявления, характерные для каждой из них.
-
Признаки, типичные для полицитемии: гепатомегалия и спленомегалия, гиперемия кожи, гипертензия, ночная потливость, головная боль, кожный зуд, диплопия, нечеткость зрения, онемение и жжение в ступнях, распирание и тяжесть в левом подреберье.


картина крови при тромбоцитемии

общая клиника лейкозов
Диагностика
Симптоматика МПЗ — основание для назначения пациенту диагностических процедур, позволяющих подтвердить или опровергнуть наличие процесса, а также выяснить, в какой именно форме протекает патология органов кроветворения.
Обследование начинают с опроса и сбора анамнеза. Врачи уточняют, какой образ жизни ведет больной, имеет ли пагубные пристрастия, какие заболевания перенес и чем лечился. Осмотр пациента – определение общего состояния и выявление признаков, которые обычно отсутствуют у здоровых людей.

Лабораторная диагностика МПЗ заключается в проведении целого ряда исследований и испытаний:
- Гемограмма — подсчет лейкоцитарной формулы, определение количества эритроцитов, тромбоцитов, уровня гемоглобина, гематокрит.
- Микроскопия мазка периферической крови — обнаружение бластных форм.
- БАК — определение функционального состояния печени и других внутренних органов.
- Миелограмма – результат микроскопии мазка пунктата костного мозга, отражающий качественный и количественный состав ядросодержащих клеток миелоидной ткани.

пункция КМ для миелограммы
Помимо лабораторной диагностики для постановки диагноза необходимы результаты инструментальных исследований. Больным проводят УЗИ брюшной полости для определения степени гепатоспленомегалии. В диагностически сложных случаях их направляют на томографическое исследование.
Лечение
Онкогематологи назначают лечение своим больным по результатам диагностических исследований. Существуют стандартные терапевтические методики, которые применяют при различных видах МПЗ. Если у пациента обнаружена начальная стадия процесса, когда еще отсутствуют клинические признаки, за ним устанавливают динамическое наблюдение. При появлении первых признаков патологии переходят непосредственно к лечению.

Каждому больному подбирается индивидуальная лечебная методика в соответствии с его состоянием и степенью выраженности имеющихся нарушений.

трасплантация КМ – наиболее радикальная, но и потенциально действенная методика при удачном исходе
После проведения полного лечебного курса наступает период реабилитации. Больной должен находится под постоянным наблюдением доктора и строго выполнять все его предписания, позволяющие организму быстрее восстановиться.
- правильное, сбалансированное питание с ограничением жирных, соленых, острых блюд и полным исключением алкоголя, курения;
- долгие пешие прогулки на свежем воздухе, желательно около водоема;
- исключение чрезмерного физического перенапряжения;
- соблюдение режима дня – полноценный сон, чередование труда и отдыха.
Миелопролиферативное заболевание — рецидивирующий процесс, способный обостриться в любое время. Именно поэтому всем пациентам необходимо регулярно посещать лечащего врача и проходить диагностические исследования с профилактической целью.
Прогноз МПЗ считается благоприятным только в случае успешной трансплантации костного мозга, которая разрешена не всем больным. Хронические формы переносятся легче острых. Продолжительность жизни пациентов в этом случае составляет 5–7 лет при условии получения комплексной терапии. Если у больных обнаружены метастазы, прогноз становится неутешительным — они погибают в течение 6 месяцев.
Видео: лекция об опыте лечения ХМПЗ
Общие сведения
Патогенез
Истинная полицитемия – это клональная неопластическая болезнь, основой которой является процесс трансформации кроветворной стволовой клетки. Вследствие дефекта такой клетки происходит соматическая мутация в гене янускиназы рецепторов цитокинов. Это приводит к пролиферации миелоидных ростков кроветворения, в основном эритроцитарного, что повышает риск развития тромбозов и тромбоэмболий.
Продолжительная пролиферация гемопоэтических клеток провоцирует развитие фиброза и замещение костного мозга коллагеновыми волокнами, вследствие чего развивается вторичный постполицитемический миелофиброз. В некоторых случаях прогрессирование болезни продолжается, и она переходит в фазу бластной трансформации.
Характерным симптомом заболевания является наличие скоплений полиморфных мегакариоцитов, как небольших, так и гигантских.
В ходе развития заболевания отмечается повышение массы циркулирующих эритроцитов, увеличивается гематокрит, становится выраженной вязкость крови, существенно возрастает уровень гемоглобина. Вследствие перечисленных факторов и тромбоцитоза нарушается микроциркуляция и происходят тромбоэмболические осложнения. К процессу присоединяется миелоидная метаплазия селезенки.
У достаточно существенной части больных на стадии миелофиброза определяются хромосомные аномалии.
Классификация
Отмечается две формы полицитемии – истинная и относительная.
Истинная эритремия, в свою очередь, может быть первичной и вторичной.
Относительная форма болезни отмечается, если уровень эритроцитов нормальный, но понижается объем плазмы. Это состояние также называют ложной или стрессовой полицитемией.
Причины
До сих пор причины эритроцитоза точно не определены. В настоящее время ученые подтвердили, что причины развития заболевания связаны с наследственным фактором.
Кроме того, спровоцировать развитие этой болезни могут такие внешние факторы:
- Воздействие на организм токсических веществ — лаков, красок, химических инсектицидов.
- Продолжительный прием некоторых лекарств – антибиотиков, препаратов золота.
- Перенесенный туберкулез, некоторые вирусные заболевания.
- Влияние радиоактивного излучения.
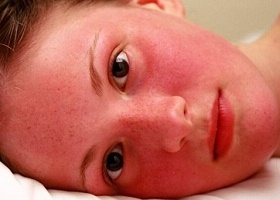
Симптомы полицитемии
По мере развития заболевания симптомы болезни Вакеза видоизменяются. Симптомы истинной полицитемии зависят от стадии заболевания. Медики выделяют четыре стадии болезни, отображающие те патологические изменения, которые происходят в селезенке и в костном мозге.
- Первая стадия – начальная. Она может продолжаться пять и больше лет. В этот период отмечается умеренный эритроцитоз и умеренная плетора, селезенка на этой стадии не пальпируется. В костном мозге отмечается панмиелоз. Есть невысокая вероятность сосудистых и тромботических осложнений. Внешне проявляется акроцианоз, плетора, эритромелалгии (парестезии и жжение в кончиках пальцев), зуд кожи после мытья. Когда увеличивается общий объем циркулирующей крови, развивается артериальная гипертензия. При этом лечение гипотензивными средствами людей, которые ранее не страдали гипертензией, является неэффективным. Постепенно развиваются проявления церебрального атеросклероза, ишемической болезни сердца. Больные жалуются на головные боли, болезненность в пальцах при движении.
- Вторая стадия – развернутая, эритремическая. Может отмечаться на протяжении 10-15 лет. Если эта стадия протекает без миелоидной метаплазии селезенки, то у больного нарушается общее состояние, отмечается выраженная плетора, панмиелоз. Высока вероятность тромботических осложнений – некрозовкончиков пальцев, инсульта, инфаркта. Больного могут беспокоить боли в костях, руках и ногах. Если у больного отмечается миелоидная метаплазия селезенки, наблюдаются гепатоспленомегалия и панмиелоз. Умеренно выражена плетора, повышена кровоточивость. Вероятны тромботические осложнения. Кожные покровы становятся багрово-синими, беспокоят жгучие боли в кончиках пальца, мочках ушей. Возможно нарушение чувствительности. Могут беспокоить боли в подреберье вследствие увеличения печени и селезенки, вероятна кровоточивость десен, боли в суставах, язвенные нарушения ЖКТ.
- Третья стадия – анемическая. Это терминальный период, в котором развивается анемический синдром, выраженный миелофиброз. Увеличивается печень, селезенка. Нарастает коллагеновый миелофиброз в костном мозге. Нарастают кахексия и спленомегалия, отмечаются проявления плеторы, а также осложнения, спровоцированные тромбозом. Происходят кровоизлияния в разных органах, усугубляется выраженность общих симптомов.
Таким образом, истинная эритремия может провоцировать следующие симптомы:
Анализы и диагностика
Для установления диагноза проводят лабораторный анализ крови и дополнительные обследования (УЗИ, КТ). Чтобы правильно установить диагноз, врачи оценивают показатели анализа крови. Эритремия определяется, если в наличии есть определенные показатели, основные и дополнительные. Подозрение на эритремию возникает, если в общем анализе крови есть отклонения, в частности, повышен гемоглобин (больше 18,5 г/дл у мужчин, больше 16,5 г/дл у женщин). Обращается внимание и на ряд других показателей (эритроцитоз, тромбоцитоз, иногда – панцитоз).
Главными критериями для установления такого диагноза являются:
- Увеличение количества эритроцитов (у мужчин — выше 36 мл/кг, у женщин — выше 32 мл/кг).
- Нормальное насыщение кислородом артериальной крови (более 92%).
- Спленомегалия.
Дополнительными критериями являются:
- Лейкоцитоз (выше 12 х 109/л без признаков инфекции).
- Тромбоцитоз (выше 400 х 109/л).
- Повышенный уровень витамина В12 (выше 900 пг/мл).
- Активность щелочной фосфатазы.
Анализируются также другие лабораторные показатели. Для подтверждения диагноза оценивают гистологическую картину головного мозга.
Кроме того, важное значение при установлении диагноза имеют такие факторы:
- Характерная внешность пациента – наличие специфической окраски кожи и слизистых.
- Увеличенная селезенка, печень.
- Склонность к образованию тромбов.
- Наличие общих симптомов, характерных для истинной полицитемии.
Дифференциальную диагностику проводят с вторичными эритроцитозами.
Лечение полицитемии
Лечение проводят так, чтобы снизить вязкость крови у больного и максимально уменьшить риск серьезных осложнений – образования тромбов и кровотечений.
Что такое истинная полицитемия?
Истинная полицитемия (также называемая эритремия, первичная полицитемия, болезнь Вакеза) — это редкое хроническое заболевание, связанное с миелопролиферацией (т.е. перепроизводством клеток крови в костном мозге). При заболевание повышается перепроизводство эритроцитов (в основном), а также лейкоцитов и тромбоцитов.
Поскольку происходит чрезмерное перепроизводство эритроцитов в костном мозге, это приводит к аномально большому количеству циркулирующих эритроцитов (красных кровяных телец) в крови. Следовательно, кровь при болезни Вакеза сгущается и увеличивается в объеме, это состояние называется гипервязкостью. Густая кровь может не проходить через меньшие кровеносные сосуды должным образом.
У лиц с истинной полицитемией могут возникать различные симптомы, включая неспецифические симптомы, такие как головные боли, усталость, слабость, головокружение или зуд кожи; увеличение селезенки (спленомегалия); различные желудочно-кишечные проблемы; и риск образования тромбов, препятствующие притоку крови к жизненно важным органам. Более 90% людей с болезнью Вакеза имеют мутации гена JAK2. Точная роль, которую играет мутация в развитии истинной полицитемии, еще не известна.
Эритремия встречается с частотой около 2 случаев на каждые 100 000 человек. Средний возраст, в котором диагностируется истинная полицитемия, равен 60 годам; однако иногда заболевание также развивается у больных в возрасте до 40 лет. В возрастной группе старше 60 лет эритремией болеют больше мужчин, чем женщин, но в возрасте до 40 лет заболевание чаще развивается у женщин.
Признаки и симптомы

Истинная полицитемия может протекать бессимптомно в течение нескольких лет. Самые ранние симптомы обычно включают:
- слабость;
- утомляемость;
- головную боль;
- предобморочное состояние;
- одышку;
- ночные поты;
- зуд после душа или ванны.
У больных может наблюдаться нарушение зрения, а перед глазами появляются слепые пятна или вспышки света (глазная мигрень).
У пациентов может развиваться кровотечение из пищеварительного тракта или десен, а также более интенсивное, чем обычно, кровотечение после небольших порезов.
Кожа, особенно на лице, может краснеть. У человека может появляться зуд по всему телу, особенно после ванны или душа. У пациентов может наблюдаться покраснение и жжение кистей и стоп (эритромелалгия). Реже может ощущаться боль в костях.
Иногда первые симптомы вызваны появлением кровяного сгустка. Повышение числа эритроцитов при истинной полицитемии приводит к сгущению крови и повышению риска образования тромбов по сравнению с нормой. Сгусток может сформироваться практически в любом кровеносном сосуде, включая сосуды, расположенные в руках, ногах (вызывая тромбоз глубоких вен), сердце (вызывая инфаркт миокарда), головном мозге (вызывая инсульт) и легких (вызывая тромбоэмболию легочной артерии). Сгустки крови также могут закупоривать кровеносные сосуды, которые отводят кровь от печени (синдром Бадда-Киари), особенно у молодых женщин.
У некоторых больных увеличивается количество тромбоцитов (клеткоподобных частиц в крови, помогающих организму формировать сгустки крови [тромбоцитемия]). Казалось бы, повышение числа тромбоцитов должно вызывать чрезмерное свертывание крови, однако на самом деле очень высокое число тромбоцитов при истинной полицитемии может привести к кровотечению, вызванному поражением других компонентов системы свертывания крови в организме.
Иногда может развиваться дефицит железа.
Может происходить увеличение размеров печени и селезенки, поскольку оба этих органа начинают вырабатывать клетки крови. Из-за увеличения печени и селезенки может появляться ощущение переполнения желудка. Если сгусток крови появляется в кровеносных сосудах печени или селезенки, вызывая гибель участков этих органов, у человека может возникать внезапная резко усиливающаяся боль.
Избыток эритроцитов может привести к развитию язвы желудка, подагры и камней в почках. В редких случаях болезнь Вакеза прогрессирует в лейкоз.
Причины
Истинная полицитемия вызвана злокачественным изменением генетического материала (ДНК) в пределах одной клетки костного мозга (клональное расстройство). Костный мозг — это мягкий, губчатый материал, который находится внутри кости, где происходит образование большинства клеток крови. Основная причина, по которой происходит это злокачественное изменение, неизвестна.
Это изменение приобретается в том смысле, что оно происходит после зачатия и не наследуется. Исследователи определили, что примерно у 90 процентов людей с истинной полицитемией есть вариации в гене JAK2. Гены предоставляют инструкции для создания белков, которые играют важную роль во многих функциях организма. Когда происходит мутация гена, белковый продукт может стать дефектным. В зависимости от функций конкретного белка это может повлиять на многие системы органов организма.
Кроме того, у некоторых пациентов с болезнью Вакеза были обнаружены дополнительные мутации в гене кальретикулина (CALR) и в других генах. Эти мутации приводят к устойчивой активации киназы JAK2, которая вызывает избыточную выработку эритроцитов.
В некоторых случаях селезенка и печень также начинают вырабатывать клетки крови. В конечном итоге у небольшой доли пациентов в костном мозге развивается миелофиброз с рубцовой тканью, и снижается его способность к выработке клеток крови.
Эпидемиология
Ежегодная заболеваемость оценивается примерно в 1/36 000-1/100 000, а распространенность — в 1/3300. Болезнь Вакеза встречается в любом возрасте, но чаще всего в возрасте 40-60 лет.
Диагностика
Истинную полицитемию можно обнаружить при проведении общего анализа крови в связи с другим заболеванием еще до возникновения каких-либо симптомов. Наблюдается чрезмерное повышение эритроцитов, уровня белка, отвечающего за транспорт кислорода в эритроцитах (гемоглобина), а также процентного содержания эритроцитов в общем объеме крови (гематокрит). Количество тромбоцитов и лейкоцитов также может быть повышенным.
Врачи могут рассматривать вероятность истинной полицитемии при существенном повышении гематокрита. Тем не менее, диагноз не может базироваться только на результатах гематокрита. Обнаружив повышенный уровень эритроцитов (полицитемию) в организме пациента, врачи должны определить тип заболевания — истинная полицитемия или полицитемия, вызванная каким-то другим состоянием (вторичный эритроцитоз). Иногда отличить истинную полицитемию от вторичного эритроцитоза помогает медицинский анамнез и осмотр, но обычно требуются дополнительные исследования.
Также может измеряться уровень содержащегося в крови эритропоэтина — гормона, который воздействует на костный мозг, стимулируя выработку эритроцитов. При истинной полицитемии уровень эритропоэтина, как правило, оказывается чрезвычайно низким, а при вторичном эритроцитозе часто (но не всегда) остается в норме или повышается.
Пациенту назначают тест на наличие мутаций гена JAK2. Если указанные мутации не обнаружены, исследуются мутации CALR.
Лечение
При лечении большинство больных с истинной полицитемией живут десятки лет. У около 15% пациентов развивается миелофиброз, а у небольшой доли может развиться острый лейкоз.
Лечение не устраняет истинную полицитемию, но может сдерживать течение заболевания и снизить вероятность возникновения осложнений, таких как формирование сгустков крови. Лечение нацелено на понижение количества эритроцитов. В большинстве случаев кровь выводится из организма в ходе процедуры под названием флеботомия, аналогично как при заборе донорской крови. Около полулитра крови извлекается каждые два дня до тех пор, пока гематокрит не достигнет нормального уровня. Затем кровь извлекается, к примеру, один раз в 1-3 месяца (по мере необходимости) для поддержания гематокрита на нормальном уровне.
Аспирин может помочь облегчить симптомы, связанные с высоким числом тромбоцитов, такие как мигрени, которые влияют на зрение, а также жгучую боль и покраснение кистей и стоп. Однако не доказано, что аспирин снижает риск образования тромбов в случае истинной полицитемии, при этом он не приносит пользы людям, у которых симптомы отсутствуют.
Пациентам, у которых после флеботомии симптомы сохраняются, могут требоваться другие виды лечения. Таким больным можно назначать руксолитиниб — препарат, ингибирующий активность JAK2, или другие препараты, включая пэгилированный интерферон альфа-2b, анагрелид или гидроксимочевину.
Трансплантация костного мозга является единственным средством для излечения истинной полицитемии, но рекомендуется только при развитии осложнений истинной полицитемии — миелофиброза и недостаточности костного мозга. Трансплантация костного мозга обычно назначается при развитии признаков миелофиброза в костном мозге.
Другие лекарственные препараты могут помогать контролировать выраженность некоторых из симптомов. Например, антигистаминные препараты, селективные ингибиторы обратного захвата серотонина (СИОЗС) или светотерапия ПУФА помогают облегчить зуд.
Прогноз
Долгосрочная перспектива для лиц, страдающих от болезни Вакеза, может зависеть от реакции на лечение. Наиболее опасным симптомом истинной полицитемии является вероятность тромботического события, которое может привести к сердечному приступу или инсульту. Существует также небольшая вероятность того, что истинная полицитемия может привести к развитию лейкемии. Однако при правильном лечении эти симптомы не оказывают существенного влияния на ожидаемую продолжительность жизни человека с болезнью Вакеза.
Другим осложнением, которое может повлиять на прогноз, является неприятный зуд, который может возникнуть как часть заболевания. Однако многие люди могут контролировать зуд с помощью лекарств, перечисленных выше.
Ph-негативные миелопролиферативные заболевания (истинная полицитемия
эссенциальная тромбоцитемия
первичный миелофиброз) у взрослых
- Национальное гематологическое общество European Hematology Association The American Society of Hematology European Society of Pathology European Association for Haematopathology Российское общество патологоанатомов Российское онкогематологическое общество
Оглавление
- Ключевые слова
- Список сокращений
- Термины и определения
- 1. Краткая информация
- 2. Диагностика
- 3. Лечение
- 4. Реабилитация
- 5. Профилактика и диспансерное наблюдение
- 6. Дополнительная информация, влияющая на течение и исход заболевания
- Критерии оценки качества медицинской помощи
- Список литературы
- Приложение А1. Состав рабочей группы
- Приложение А2. Методология разработки клинических рекомендаций
- Приложение А3. Связанные документы
- Приложение Б. Алгоритмы ведения пациента
- Приложение В. Информация для пациентов
- Приложение Г.
Ключевые слова
Список сокращений
Алло-ТГСК – аллогенная трансплантация гемопоэтических стволовых клеток
Ауто-ТГСК – аутологичная трансплантация гемопоэтических стволовых клеток
БФ – бластная фаза
ВОЗ – Всемирная организация здравоохранения
ИП – истинная полицитемия
КТ – компьютерная томография
МДС – миелодиспластический синдром
МПЗ – миелопролиферативное заболевание
МПЗн – миелопролиферативное заболевание неклассифицированное
МРТ – магнитно-резонансная томография
НМГ – низкомолекулярный гепарин
ОВ – общая выживаемость
ОМЛ – острый миелоидный лейкоз
ПМФ – первичный миелофиброз
Пост-ИП МФ – постполицитемический миелофиброз
Пост-ЭТ МФ – посттромбоцитемический миелофиброз
ПЦР – полимеразная цепная реакция
УЗИ – ультразвуковое исследование
ХМЛ – хронический миелоидный лейкоз
ХФ – хроническая фаза
ЭТ – эссенциальная тромбоцитемия
P32 – радиоактивный фосфор
DIPSS (Dynamic International Prognostic Scoring System) – Динамическая международная шкала оценки прогноза
ELN (European Leukemia Net) - Европейская организация по изучению и лечению лейкозов
EORTC - Европейская организация по исследованиям в области лечения рака
G-CSF (granulocyte colony stimulating factor) - гранулоцитарный колониестимулирующий фактор
IPSET-thrombosis (The International Prognostic Score for ET) – Международный Прогностический Индекс рисков тромбоза при эссенциальной тромбоцитемии
IPSS (International Prognostic Scoring System) – Международная шкала оценки прогноза
IWG-MRT (The international working group for myeloproliferative neoplasms research and treatment) - Международная рабочая группа по изучению и лечению миелопролиферативных заболеваний
NSSN (National Comprehensive Cancer Network®) - Национальная Онкологическая Сеть США
PVSG (Polycythemia Vera Study Group) – группа по изучению истинной полицитемии
PUVA (psoralen + UVA treatment) – ПУВА – терапия; ультрафиолетовая фототерапия в комбинации с псораленом
?? - препараты, включенные в перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения.
Термины и определения
Трепанобиопсия – диагностическая процедура забора образцов костного мозга путем пункции гребня подвздошной кости.
1. Краткая информация
1.1 Определение
Миелопролиферативные заболевания (МПЗ) представляют собой клональные заболевания, возникающие на уровне стволовой кроветворной клетки. МПЗ характеризуются пролиферацией одной или более клеточной линии миелопоэза в костном мозге с признаками сохранной терминальной дифференцировки, и сопровождаются изменением показателей периферической крови.
Истинная полицитемия (ИП) (синонимы: эритремия, болезнь Вакеза, истинная красная полицитемия) – клональное МПЗ, которое характеризуется пролиферацией эритроидного, гранулоцитарного, мегакариоцитарного ростков миелопоэза, с преимущественной пролиферацией эритроидного ростка кроветворения (панмиелоз), увеличением числа эритроцитов и повышением уровня гемоглобина, тромбоцитозом, лейкоцитозом в периферической крови (панцитоз), независимостью эритропоэза от нормальных механизмов регуляции. Почти все больные являются носителями мутации JAK2V617F или другой функционально сходной мутации.
Эссенциальная тромбоцитемия (синонимы: первичный тромбоцитоз, идиопатический тромбоцитоз, геморрагическая тромбоцитемия) – клональное МПЗ с неконтролируемой пролиферацией мегакариоцитов, характеризующееся повышенным числом крупных и гигантских мегакариоцитов в костном мозге, тромбоцитозом в периферической крови (>450 х 109/л), высоким риском тромбозов и/или кровотечений.
Миелопролиферативное заболевание, неклассифицируемое (МПЗн) Согласно рекомендациям ВОЗ 2008 г., данный диагноз следует использовать при наличии клинических, лабораторных и гистологических (в трепанобиоптате костного мозга) признаков МПЗ, не соответствующих какой-либо определенной нозологической форме классических Ph-негативных МПЗ. Чаще всего эта категория используется: При ранних стадиях заболевания (манифестация) – при расхождении между клиническими, лабораторными и морфологическими данными, позволяющими верифицировать ту или иную нозологическую форму МПЗ. При бластной фазе заболевания, без предшествующего анамнеза и установленного ранее диагноза миелопролиферативного заболевания. При сочетании МПЗ с воспалительными, метаболическими или опухолевыми заболеваниями, маскирующими основные признаки той или иной нозологичесой формы. МПЗ неклассифицируемое не диагностируется: при объеме трепанобиоптата костного мозга недостаточном для адекватного анализа; при отсутствии предоставленных клиницистами клинических и лабораторных данных, при наличии предшествующей терапии цитостатиками или колониестимулирующими факторами; при наличии реаранжировки генов PDGFRA, PDGFRB, FGFR1, выявлении химерного гена BCR-ABL1.
Первичный миелофиброз (ПМФ) (синонимы: хронический идиопатический миелофиброз, агногенная миелоидная метаплазия, миелосклероз с миелоидной метаплазией, сублейкемический миелоз, хронический гранулоцитарно-мегакариоцитарный миелоз) возникает de novo, характеризуется клональной пролиферацией стволовых клеток, аномальной экспрессией цитокинов, фиброзом костного мозга, гепатоспленомегалией как следствие экстрамедуллярного гемопоэза, симптомами опухолевой интоксикации, кахексией, лейкоэритробластозом в периферической крови, лейкемической прогрессией, невысокой выживаемостью.
1.2 Этиология и патогенез Ph - негативных МПЗ
Этиология МПЗ до сих пор не установлена. Ведущей гипотезой является многоэтапность возникновения заболевания, где предрасположенность к болезни реализуется под воздействием внешних факторов, повреждающих геном нормальной клетки и приводящих к ее злокачественной трансформации. Несмотря на то, что в последние годы достигнуты значительные успехи в расшифровке молекулярно-генетических механизмов Ph-негативных МПЗ, первоначальная мутация, приводящая к малигнизации гемопоэтической клетки неизвестна [1].
Открытие мутации V617F в гене JAK2 в 2005г явилось значительным шагом в понимании биологических особенностей Ph-негативных МПЗ. Практически у всех пациентов с ИП выявляется мутация гена JAK2: в 96% случаев мутация JAK2V617F (14 экзон), в 2% наблюдений мутация в экзоне 12 гена JAK2 [2, 3]. Мутация JAK2V617F выявляется при ЭТ в 55% наблюдений и присутствует примерно в 45 - 68% случаев при ПМФ. Тогда как мутация в 12 экзоне гена JAK2 практически не встречается при ЭТ и ПМФ [4,5].
Помимо мутации гена JAK2 у больных МПЗ выявляют мутации и в других генах. Мутации гена MPL встречаются в 4% наблюдений при ЭТ, в 8% наблюдений при ПМФ, и редко при ИП. Причем наиболее частые мутации MPLW515L/K в экзоне 10 [6, 7, 8]. Мутация MPLS505N выявляется как при ЭТ, так и при наследственной тромбоцитемии [9, 10]. Данные мутации не являются строго специфичными для МПЗ и имеют вторичный генез в цепи генетических событий.
Не так давно появились данные о диагностической значимости соматических мутаций в 9 экзоне гена CALR, кодирующего белок кальретикулин. Выявлены 36 разных видов мутаций в этом гене, которые приводят к образованию дефектного белка. В исследованиях in vitro клетки, экспрессирующие мутированный ген, обладали способностью цитокин-независимого роста в культуре, что вероятно связано с активацией белков STAT5. У пациентов без мутаций в генах JAK2 и MPL мутации в данном гене были выявлены в 67% случаев при ЭТ и 88% - при ПМФ. Другие авторы также выявили крайне высокую частоту мутаций гена CALR у пациентов с МПЗ (в 70-84% случаев при отсутствии мутации гена JAK2). При этом мутации CALR были обнаружены в 8% случаев при миелодиспластическом синдроме и в единичных наблюдениях при других миелоидных неоплазиях. Важно, что ни в одном случае заболеваний не миелоидной природы, мутации в данном гене не были выявлены [11,12].
Мутации в генах JAK2, MPL, CALR имеют важное диагностическое значение. Их выявление свидетельствует о клональном характере заболевания и помогает в дифференциальной диагностике ИП, ЭТ, ПМФ от ряда других миелоидных неоплазий, а также вторичных эритроцитозов и тромбоцитозов. Наряду с этим активно изучается значимость данных мутаций в прогнозе МПЗ. Несмотря на ряд проведенных исследований, не представляется возможным сделать однозначное заключение в отношении прогностической значимости аллельной нагрузки JAK2V617F при ИП, ЭТ, ПМФ. Вопрос влияния аллельной нагрузки на выживаемость или прогрессирование ИП и ЭТ с исходом в миелофиброз требует дальнейшего изучения [13, 14].
При ИП и ЭТ выявляются и другие мутации: TET2, IDH, ASXL1, DNMT3A и др. [1]. Ни одна из них не специфична для классических Ph-негативных МПЗ, а их патогенетическая значимость исследуется.
Молекулярно-генетические нарушения при Ph-негативных МПЗ приводят к активации JAK-STAT сигнального пути. Результатом этого является повышение пролиферации и увеличение количества эритроцитов, лейкоцитов и тромбоцитов периферической крови при ИП или изолированный тромбоцитоз при ЭТ. Патогенез МПЗ, в частности ПМФ, сложен и состоит из цепи событий, первичным из которых является появление патологического клона. Известно, что лейкемические моноциты и мегакариоциты активно продуцируют множество цитокинов (TGF-?, FGF, VEGF, ANG1, OPG, BMP4), избыток которых стимулирует фиброз, неоангиогенез и приводит к остеосклерозу. Наряду с этим нарушается связь стволовых клеток с микроокружением, что способствует появлению экстрамедуллярных очагов гемопоэза, прежде всего в селезенке и печени. Массивный выброс цитокинов - одна из причин возникновения симптомов опухолевой интоксикации, что приводит к значительному ухудшению качества жизни пациентов с ПМФ [15].
Клональная миелопролиферация при Ph-негативных МПЗ также может сопровождаться вторичным воспалением с изменениями стромы костного мозга и патологической выработкой цитокинов. В развитии миелофиброза, как первичного, так и вторичного, остеосклероза и ангиогенеза вовлечены трансформирующий фактор роста бета (TGF-?) миелоидных предшественников, ростовой фактор вырабатываемый тромбоцитами (PDGFR) и эндотелиальный сосудистый фактор роста (VEGF) [16]. Патологическая выработка цитокинов, хемокинов и металлопротеиназ может участвовать в патологическом межклеточном взаимодействии нейтрофилов, моноцитов и мегакариоцитов, приводя к выходу CD34+ миелоидных предшественников и эндотелиальных клеток в периферическую кровь [17, 18].
1.3 Эпидемиология
ЭТ – редкое / орфанное заболевание. Популяционные эпидемиологические данные о заболеваемости и распространенности в России отсутствуют. Литературные данные о заболеваемости по данным зарубежных регистров составляют приблизительно 1,5 - 2,53 : 100 000 населения [19]. При анализе десятилетней динамики заболеваемости в Санкт-Петербурге ежегодная первичная заболеваемость колебалась от 0,60 до 2,10 и составила в среднем 1,30 на 100 000 населения в год [20].
1.4 Кодирование по МКБ 10
D47.4 – первичный миелофиброз
D45 – истинная полицитемия
D47.3 – эссенциальная тромбоцитемия
1.5 Классификация
В соответствии с классификацией ВОЗ 2008г группа хронических МПЗ объединяет восемь нозологических форм:
Читайте также: