
Хронические лимфопролиферативные лейкоз студфайлс
–лимфопролиферативное неопластическое заболевание, характеризующееся пролиферацией и увеличением в периферической крови количества зрелых лимфоцитов на фоне лимфоцитарной инфильтрации костного мозга, лимфатических узлов, селезенки и других органов.
Клеточный субстрат хронического лимфолейкоза (ХЛЛ) представлен морфологически зрелыми лимфоцитами, в основном В-лимфоцитами (до 95%) и значительно реже – Т-лимфоцитами (около 5%).
Распространенность.Варьирует в различных географических регионах и этнических группах. Частота 2,7:100000 населения. Болеют в основном пожилые, мужчины вдвое чаще женщин. Детская заболеваемость казуистична.
Этиология.В развитии ХЛЛ наиболее очевидна роль вирусной инфекции (ретровирусов) и генетических факторов. Имеются семьи, в которых многие ее члены болеют ХЛЛ, и предрасположенность к развитию ХЛЛ прослеживается во многих поколениях. Приблизительно у 50% больных ХЛЛ обнаруживаются хромосомные аномалии, причем наиболее часто в области 12, -13, -14-й хромосом.
Патогенез.Хронический лимфолейкоз, по современным представлениям, является клональным заболеванием. Образовавшийся патологический клон клеток развивается по законам опухолевой прогрессии, но значительно медленнее и менее агрессивно, чем при остром лейкозе. Лимфоциты вначале накапливаются в лимфатических узлах и далее в других лимфоидных тканях, увеличиваются печень и селезенка, развивается прогрессирующая лимфоцитарная инфильтрация костного мозга. По мере прогрессирования заболевания нарушается гемопоэз, развиваются анемия, грануло- и тромбоцитопения, нарушается функция иммунной системы, так как лимфоциты функционально неполноценны. Развиваются различные аутоиммунные конфликты (гемолитическая анемия, васкулиты и др.), часто инфекционно-воспалительные заболевания.
Классификация.ХЛЛ подразделяют на В- и Т-формы.
По клиническому течению А.И. Воробьев выделяет следующие формы:
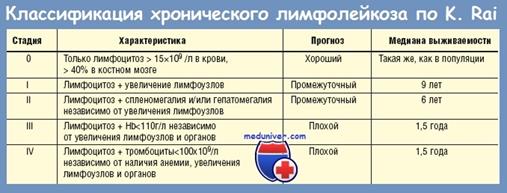
Клинические стадии ХЛЛ по Rai (1989)
0 – лимфоцитоз в периферической крови (средняя продолжительность жизни 10-12 лет),
I – лимфоцитоз и увеличение лимфатических узлов,
II – лимфоцитоз и спленомегалия, (пациенты обычно живут 4–7 лет),
III – лимфоцитоз и анемия,
IV – лимфоцитоз и тромбоцитопения.
При появлении гематологических симптомов прогноз ухудшается (средняя продолжительность жизни около 18 месяцев).
Клиническая картина.В клинике присутствуют различной степени выраженности синдромы, характерные для гемобластозов: гиперпластический, анемический, интоксикационный, иммунодефицитный, но в отличие от клинических проявлений при остром лейкозе они носят постепенный и некритический характер.
В начальном периоде заболевания больные обычно не предъявляют жалоб, общее состояние удовлетворительное. Могут беспокоить небольшая слабость, потливость. Как правило, заболевание выявляется случайно. Основными клиническими признаками на этой стадии являются увеличение лимфоузлов, лейкоцитоз и лимфоцитоз. Обычно в первую очередь увеличиваются шейные, затем подмышечные, позднее другие группы лимфатических узлов. Степень увеличения узлов может быть различной: от небольшой до весьма значительной. Как правило, лимфоузлы безболезненны, не спаяны с кожей и между собой, не нагнаиваются. В общем анализе крови характерен лейкоцитоз, не превышающий 50´10 9 /л, лимфоцитоз до 60–80%.
В периоде выраженных клинических проявлений больные жалуются на резкую слабость, снижение работоспособности, потливость, особенно по ночам, похудание, повышение температуры тела, увеличение лимфоузлов практически всех групп. При осмотре выявляются лимфаденопатия, увеличение печени и селезенки. Увеличенная селезенка плотная, поверхность ее гладкая. Спленомегалия может сопровождаться синдромом гиперспленизма. Лейкемическая инфильтрация дыхательных путей и легких проявляется кашлем, одышкой, редко кровохарканьем.
В анализе крови отмечается лейкоцитоз различной степени выраженности, обычно 50–100´10 9 /л, значительный лимфоцитоз (80–90%). Характерным признаком является появление клеток Боткина-Гумпрехта – полуразрушенных ядер лимфоцитов в ходе приготовления мазка крови. У 50% больных ХЛЛ обнаруживается нормохромная анемия, довольно часто развивается тромбоцитопения, характерно увеличение СОЭ. В биохимическом анализе крови определяются признаки гемолиза: непрямая гипербилирубинемия.
В миелограмме наблюдается выраженная лимфоидная инфильтрация вплоть до тотальной лимфатической метаплазии костного мозга.
Заключительная (терминальная) стадия ХЛЛ характеризуется резким прогрессирующим ухудшением общего состояния больных, истощением, выраженной интоксикацией, высокой температурой тела. Для этой стадии характерно развитие тяжелых осложнений, в первую очередь инфекционно-воспалительных процессов различных локализаций. Тяжелая генерализованная инфекция часто является причиной смерти больных ХЛЛ. В терминальной фазе ХЛЛ возможно развитие нейролейкемии в связи с инфильтрацией мозговых оболочек молодыми лимфоцитами.
В ОАК выявляется выраженная анемия, тромбоцитопения, обусловливающая геморрагический синдром, могут появляться бласты.
Диагностика. Международным рабочим совещанием по ХЛЛ в 1989 г. предложены следующие диагностические критерии ХЛЛ:
– абсолютное количество лимфоцитов в периферической крови > 10´10 9 /л;
– количество лимфоцитов в миелограмме составляет более 30% всех ядросодержащих клеток;
– большинство лимфоцитов в периферической крови имеют иммунологические маркеры В-лимфоцитов.
Дифференциальная диагностика.В начальной стадии ХЛЛ следует дифференцировать с заболеваниями, которые могут сопровождаться выраженной лейкемоидной реакцией лимфоцитарного типа: с туберкулезом легких и лимфатических узлов, с некоторыми вирусными инфекциями. При появлении лимфаденопатии необходимо дифференцировать с лимфогранулематозом и неходжкинскими лимфомами.
При лейкемоидных реакциях нивелирование основного воспалительного процесса нормализует картину крови. Безусловно, ведущим достоверным методом дифференциальной диагностики является миелограмма, а в случае лимфогранулематоза – особенности клинических проявлений последнего и результаты гистологического исследования пунктата лимфоузлов (специфичны клетки Березовского-Штернберга).
Лечение.На ранней стадии заболевания при стабильном лейкоцитозе 20–30´10 9 /л лечение не проводят. Показано только наблюдение, периодический (1 раз в 3–6 мес.) контроль анализа крови.
Показаниями к назначению цитостатиков являются:
появление лихорадки, похудания, потливости;
нарастание лейкоцитоза выше 50´10 9 /л;
увеличение лимфатических узлов, печени, селезенки.
Применяют следующие цитостатики:
Хлорбутин (лейкеран, хлорамбуцил) – по 5–10 мг 1–3 раза в неделю. Общая доза на курс лечения составляет 100–700 мг.
Циклофосфан – внутривенно или внутримышечно в дозе 200–400 мг ежедневно или через день. Курсовая доза в среднем 8–12 г.
Флударабин – высокоактивный препарат при ХЛЛ. Назначают в дозе 25 мг/м 2 внутривенно в течение 30 минут 5 дней подряд. Число курсов 6–10.
При опухолевом варианте ХЛЛ, прогрессирующем течении, пролимфоцитарном варианте назначается полихимиотерапия по схемам: СОР (циклофосфан + винкристин (онковир) + преднизолон), СНОР (СОР + доксорубицин).
Лучевая терапия применяется при резко выраженном увеличении лимфатических узлов и селезенки.
При спленомегалии в сочетании с гиперспленизмом, панцитопении показана спленэктомия, при гиперлейкоцитозе более 200´10 9 /л – лимфоцитоферез.
Прогноз.Выздоровления не наблюдается. Средняя продолжительность жизни составляет около 4–6 лет после начала химиотерапии. При медленном доброкачественном течении заболевания продолжительность жизни составляет более 10 лет.
Опухоль кроветворной ткани, исходящая из клеток-предшественниц миелопоэза, морфологическим субстратом которой являются дифференцирующиеся (промежуточные формы) и зрелые гранулоциты. Клон клеток, формирующийся при хроническом миелолейкозе (ХМЛ), на определенном этапе оказывается нестабильным, и это приводит к развитию тяжелой стадии – бластного криза, рефрактерного к терапии и заканчивающегося, как правило, летальным исходом.
Распространенность.Заболевание встречается с частотой 1–1,7 случая на 100000 населения, что составляет около 20% случаев гемобластозов у взрослых. Мужчины болеют чаще, чем женщины. Заболевание развивается обычно в возрасте 30–50 лет, в детском и юношеском возрасте встречается редко.
Этиология и патогенез.В развитии заболевания доказана роль:
– ионизирующей радиации (среди пострадавших от атомных бомбардировок японских городов Хиросимы и Нагасаки лейкоз развился через 11 лет в 30% случаев; среди женщин, которые получали лучевую терапию по поводу рака шейки матки, ХМЛ диагностирован через 9 лет в 30% случаев);
Химические вещества как этиологические факторы ХМЛ пока достоверно не идентифицированы.
Классификация (Аthens, 1993).
Клинические варианты:
● типичный ХМЛ (с филадельфийской хромосомой);
● атипичный ХМЛ (без филадельфийской хромосомы);
Фазы клинического течения:
● хроническая стабильная фаза (развернутая стадия);
● фаза бластного криза (терминальная стадия).
| | | следующая лекция ==> | |
| Дифференциальный диагноз | | | Клиническая картина. В начальной фазе заболевания многие больные активных жалоб не предъявляют |
Нам важно ваше мнение! Был ли полезен опубликованный материал? Да | Нет
Хронический лимфолейкоз (ХЛЛ) – клональное лимфопролиферативное неопластическое заболевание, характеризующееся пролиферацией и увеличением в периферической крови количества зрелых лимфоцитов на фоне лимфоцитарной инфильтрации костного мозга, лимфатических узлов, селезенки и других органов.
Эпидемиология: около 30% всех лейкозов; заболеваемость 2,7 на 100 тыс.; болеют в основном пожилые; в 95% случаев клеточный субстрат представлен В-лимфоцитами, в 5% - Т-лимфоцитами
Этиология ХЛЛ:
1) ретровирусы – их роль в развитии ХЛЛ сегодня уже очевидна
2) наследственная предрасположенность (хромосомные аномалии, обнаруживаются в 100% случаев; наиболее характерны трисомия 12, делеция длинного плеча 13 хромосомы, избыток генетического материала в 14 хромосоме и др.)
В настоящее время развитие ХЛЛ не ассоциируется с воздействием ионизирующего излучения, лекарств, химических веществ.

Патогенез В-клеточного ХЛЛ (как наиболее частой формы):
1. Возникновение патологического клона клеток-предшественниц, дифференцирующихся только по В-лимфоцитарному пути, вследствие воздействия вирусов или спонтанных генетических мутаций
2. Развитие патологического клона клеток по законам опухолевой прогрессии, но значительно медленнее и менее агрессивно, чем при остром лейкозе, с преобладанием зрелых лимфоцитов, накапливающихся вначале в лимфатических узлах, далее – в лимфоидных тканях, печени, селезенки, костном мозге.
3. Прогрессирующая лимфоцитарная инфильтрация костного мозга, приводящая к нарушениям гемопоэза (анемии, гранулоцитопении, тромбоцитопении), продукции иммуноглобулинов, возникновению аутоиммунных конфликтов (из-за неполноценности лимфоцитов патологического клона).
Клинические проявления и диагностика ХЛЛ:
а) начальный период:
- общее состояние удовлетворительное, жалобы на небольшую слабость, потливость, частые простудные заболевания
- небольшое увеличение л.у. (в первую очередь – шейных, затем подмышечных, остальные л.у. увеличиваются в развернутой стадии заболевания); л.у. эластично-тестотваты, безболезненны, не спаяны с кожей и между собой, не изъязвляются и не нагнаиваются
Диагностика: лейкоцитоз 10-30*109/л, не имеющий тенденции к увеличению; абсолютный лимфоцитоз до 60-80%
б) период выраженных клинических проявлений:
- жалобы на резко выраженную общую слабость, снижение работоспособности, значительную потливость, особенно ночью, похудание, повышение температуры тела
- выраженная лимфаденопатия (увеличены практически все группы периферических л.у. в различной степени – от горошины до куринового яйца); л.у. эластично-тестоватые, безболезненные, не спаяны между собой и с кожей
- неспецифические поражения кожи (обострения ранее существовавших болезней – экземы, псориаза; опоясывающий герпес, иногда генерализованного характера; крапивница, нейродермит; грибковые поражения)
- проявления лейкемической инфильтрации внутренних органов:
- значительно увеличенная селезенка (но меньше, чем при ХМЛ), плотная селезенка с гладкой поверхностью, реже – увеличенная печень с загругленным плотноватым краем и гладкой поверхностью
- язвенное поражение желудка, ДПК, желудочно-кишечные кровотечения, синдром мальабсорбции (из-за лейкемической инфильтрации слизистой ЖКТ)
- очаговые (клинически не проявляются) или диффузные диссеменированные (клинически – одышка, кашель, кровохарканье, крепитация над всей поверхностью легких) инфильтраты в легких; фибринозный или экссудативный плеврит; частые инфекционно-воспалительные заболевания органов дыхания
- очаговые поражения миокарда (клинически не проявляются) или миокардиодистрофия; повышение проницаемости сосудов различного калибра
- поражение почек с умеренной протеинурией, реже микрогематурией без нарушения их функции
- приапизм у мужчин (длительная и болезненная эрекция из-за лейкемической инфильтрации кавернозных тел)
- нейролейкемия с развитием менингита, менингоэнцефалита, параличей и даже комы
Диагностика:
1) ОАК: лейкоцитоз (50-200*109/л), резкий лимфоцитоз периферической хрови (10*109/л и более при норме
Дата добавления: 2019-11-25 ; просмотров: 60 ;
ХЛЛ - это костномозговой опухолевый процесс лимфоидной ткани, субстратом которого являются зрелые лимфоциты. Это доброкачественная опухоль иммуннокомпетентной ткани, которая развивается из клетки-предшественницы лимфоцитопоэза. Чаще (более 90% всех случаев) встречается В-форма ХЛЛ, клеточным субстратом которого являются В-лимфоциты. Реже наблюдается Т-форма ХЛЛ, субстрат его составляет Т-популяция лимфоцитов.
Среди лейкозов ХЛЛ занимает второе место после острых. Чаще это заболевание встречается среди обитателей стран Европы, США, Канады. Намного реже оно наблюдается среди обитателей Юго-восточной и Восточной Азии.
Болеют, в основном, лица в возрасте выше 50 лет. Заболевание в более молодом возрасте встречается редко, совсем не наблюдается среди детей и юношей. Мужчины болеют значительно чаще женщин.
Этиология и патогенез
ХЛЛ, как и все гемобластози, имеют мутационное происхождение - опухоль развивается из одной клетки-предшественницы. У больных В-клеточной формой ХЛЛ оказываются хромосомные изменения в виде трисомии 12 пар хромосом и транслокации ряда хромосом (2, 4, 7, 11, 14), которые подтверждают клональный характер заболевания.
Роль внешних факторов, в том числе и роль радиации, не обнаружена. Определенную роль играют вирусы. В возникновении ХЛЛ большую роль играет фактор наследственности. В пользу роли эндогенных генетических факторов говорят:
1. Преимущество среди заболевших лиц старших возрастных групп и лиц мужского пола.
2. Оно чаще встречается среди определенных национально-этнических групп населения (евреи).
3. Случаи заболевания ХЛЛ или ХЛЛ и лимфосаркомой у нескольких членов одной семьи.
4. Высокая заболеваемость ХЛЛ среди лиц с разными врожденными дефектами соединительной ткани.
5. Высший процент заболеваемости ХЛЛ среди людей с наследственным иммунодефицитом.
При ХЛЛ происходит лимфоидная инфильтрация костного мозга, лимфатических узлов, селезенки, печени, которая приводит к увеличению этих органов и тканей, иногда значительному. Продолжительность жизни лимфоцитов увеличивается в 5 раз по сравнению с нормой, а продукция их возрастает примерно в 10 раз.
В-лимфоциты при ХЛЛ не дифференцируются к образованию клеток, которые производят иммуноглобулины. Возникает функциональная неполноценность лимфоцитов, нарушается антителообразование. В крови многих больных снижен титр комплимента, возникает ослабление иммунитета, следствием чего является тяжело протекающие инфекционные заболевания, в том числе и вирусные, которые могут стать причиной смерти больных.
Вместе с этим нарушения иммуннологического гомеостаза приводят к аутоиммунным осложнениям в виде аутоиммунной гемолитической анемии, протекающей с гемолитическими кризами, аутоиммунных тромбо- и гранулоцитопений.
Клиническая картина заболевания чрезвычайно разнообразна, что зависит от стадии заболевания и клинико-гематологического варианта болезни.
В большинстве случаев заболевание начинается постепенно и длительно протекает латентно. Многие больные в этот период не знают о том, что они больны. У них отмечается лишь небольшая астенизация. О заболевании они могут узнать случайно, после проведенного по какого-либо поводу анализа крови. Некоторые больные обращаются к врачу в связи с выявленным ими увеличением лимфоузлов - чаще шейных. Лимфатические узлы тестоватой консистенции, подвижные, безболезненные, не спаянные между собой и окруженные тканями. Они могут быть разной величины, однако вначале они чаще небольшие. Это первая - начальная стадия заболевания, длительность которой от нескольких месяцев до нескольких лет. Нередко трудно определить начало заболевания. Реже начальный период заболевания не диагностируется, а к врачу больной обращается, когда наступает ІІ стадия.
ІІ стадия - стадия развернутых клинических проявлений. В этой стадии изменяется самочувствие больного - появляются признаки интоксикации. Больные жалуются на общую слабость, снижение работоспособности, потерю аппетита, потливость. Периодически может повышаться температура тела, чаще до субфебрильного уровня. Отмечается генерализованное увеличение лимфатических узлов - увеличиваются подмышечные, паховые лимфоузлы, потом медиастенальные, брыжеечные и внебрюшинные. Лимфоидной инфильтрации могут подвергнуться околоушные и слезные железы, которая вызывает сухость слизистых оболочек полости рта и глаз (симптом Микулича). Лимфатические узлы могут достигать больших размеров, образовывая конгломераты, которые в некоторых случаях вызывают сжатие, например, вен портальной системы. Печень и селезенка увеличены, однако чаще не достигают таких размеров, как при ХМЛ. Возможно развитие специфической лимфоидной инфильтрации в легочной ткани. Часто наблюдается поражение пищеварительного тракта, которое связано с возникновением специфического инфильтрата в слизистом и подслизистом слоях кишок, богатых лимфоидной тканью. Возникают метеоризм, запоры, часто понос. Со стороны сердца определяются признаки миокардиодистрофии. Со стороны почек может возникнуть мочекаменная болезнь из-за распада большого количества лейкоцитов с проникновением в кровь солей мочевой кислоты. Нередко разнообразные неспецифические высыпания на коже в виде эритем, опоясывающего лишая, высыпаний, напоминающих пузырчатку. Реже наблюдается особенная кожная форма ХЛЛ - так называемая Т-клеточная, когда кожа поражается специфическим инфильтратом. В этой стадии развивается анемия.
ІІІ - терминальная стадия - характеризуется резким ухудшением общего состояния, повышением температуры, снижением массы тела больного, ростом анемии, геморрагическим синдромом. На этой стадии происходит распространение лимфоидной инфильтрации (тотальная лимфоидная инфильтрация костного мозга, лимфоидные инфильтраты в легких, плевре, ЦНС, кишечнике, миндалинах и др.).
Гематологическая картина характеризуется, прежде всего, высоким гиперлейкоцитозом, хотя он может быть незначительным, а в ряде случаев количество лейкоцитов может сохраняться нормальным. Отмечается абсолютный и относительный лимфоцитоз. Количество лимфоцитов может быть 50-70%, нередко 80-90% и выше. В начальной стадии заболевания лейкоцитоз и количество лимфоцитов увеличены незначительно, а по мере прогрессирования заболевания, особенно в период обострения, их количество растет. Лимфоциты представлены зрелыми формами, количество молодых лимфоцитов - промиелоцитов не превышает 5-10%. Их количество увеличивается при обострении заболевания и в терминальной стадии. В мазке крови находят тени Гумпрехта - разрушенные ядра лимфоцитов, особенно пролимфоцитов, что объясняется неустойчивостью их и разрушением при приготовлении мазка. На терминальной стадии может произойти бластный криз с выходом в кровь лимфобластов. В лимфоцитах при цитохимическом исследовании оказывается повышенное содержание гликогена, иногда в виде гранул. Отмечается относительная и абсолютная гранулоцитопения.
На начальных стадиях заболевания анемия, конечно, не наблюдается. По мере прогрессирования заболевания анемия возникает и нарастает в связи с угнетением нормального кроветворения растущей опухолевой массой. Она обусловлена и уменьшением продолжительности жизни эритроцитов. Кроме того, возникает осложнение заболевания анемией аутоиммунного происхождения. Последняя является нормохромной и характеризуется ретикулоцитозом, позитивной пробой Кумбса.
Тромбоцитопения и гранулоцитопения развиваются при выраженной опухолевой инфильтрации костного мозга, а также в случаях возникновения тромбоцитопении и гранулоцитопении аутоиммунного происхождения. У многих больных оказывается гипогаммаглобулинемия. В миелограмме выявляется преимущество лимфоцитов - в начальных стадиях 40-50%, в развернутой стадии - 50-70%, в терминальной стадии - 70-90%.
Различают несколько форм ХЛЛ (А.И.Воробъев, 1985):
I. Свободнопрогрессирующая, доброкачественная форма. Она характеризуется длительным течением заболевания, с сохранением удовлетворительного состояния больного, в начале - небольшим увеличением лимфатических узлов, медленным увеличением лейкоцитоза и лимфоцитоза, длительным отсутствием анемии, не частыми осложнениями.
2. Быстропрогрессирующая форма - быстрое ухудшение состояния больного, быстрое увеличение лимфоидной инфильтрации, анемия; выражен лейкоцитоз за счет лимфоцитоза.
3. Опухолевая - значительное увеличение всех групп лимфатических узлов. Может быть умеренное или значительное увеличение селезенки. Лейкоцитоз, лимфоцитоз умеренно выраженные. Мало изменяется эритро- и тромбоцитопоэз.
4. Спленомегалическая. Характеризуется значительным увеличением селезенки при небольшом увеличении лимфатических узлов. Лейкоцитоз и лимфоцитоз умеренно выражены.
5. Костномозговая. Она характеризуется выраженной диффузной инфильтрацией костного мозга и растущей цитопенией. Она протекает без увеличения лимфатических узлов и селезенки.
6. Т-клеточная. Характеризуется выраженной спленомегалией, поражением печени, непостоянным увеличением лимфатических узлов и поражением кожи. Последнее проявляется эритематозно-десквамативными изменениями. Чаще отмечается небольшой лейкоцитоз, анемия. Течение неблагоприятное.
7. Волосоклеточный. Редкая форма ХЛЛ. Характеризуется значительным увеличением селезенки, реже - печени, небольшим увеличением лимфатических узлов или чаще отсутствием их увеличения, лимфоцитозом, панцитопенией. Эта форма названа так потому, что среди лимфоцитов оказываются "волосяные" клетки, которые имеют большие размеры, их цитоплазма избыточна, с тонкими отростками, которые напоминают волосы. Для них типична диффузная реакция на кислую фосфатазу. Течение заболевания разнообразное.
Диагноз и дифференциальный диагноз
В большинстве случаев постановка диагноза не вызывает трудности. Диагноз определяется на основе выявления увеличенных характерных для лимфолейкоза лимфатических узлов, которые часто сопровождаются увеличением селезенки, реже - печени, выраженным лимфоцитозом, часто лейкоцитозом. Дифференциальный диагноз проводится с внекостномозговыми гемобластозами, также протекающими совместно с лимфоаденопатией - лимфогрануломатозом, лимфосаркомами. В отличие от ХЛЛ характеристики лимфоузлов при этих заболеваниях (плотность, спаянность лимфоузлов между собой, прорастание в окружающих ткани, последовательность их втяжения в процесс) большую роль играют анализы крови (отсутствие выраженного лейкоцитоза, лимфоцитопении и др.), а также отсутствие лимфоидной инфильтрации костного мозга. Решающую роль в диагностике этих заболеваний играют результаты исследования биоптата, взятого из лимфатического узла.
Иногда приходится проводить дифференциальный диагноз с лимфоцитарной лейкемоидной реакцией, инфекционным мононуклеозом, а также агранулоцитозом. В последнем случае дифференциальный диагноз бывает необходим в связи с высоким лимфоцитозом, который констатируется при обоих заболеваниях. Однако при ХЛЛ лимфоцитоз абсолютен, а при агранулоцитозе он относителен.
Течение заболевание и прогноз
ХЛЛ относится к числу доброкачественно протекающих лейкозов. Однако, наряду с доброкачественнопротекающим лейкозом, существует и быстропрогрессирующий. Продолжительность жизни колеблется в широких пределах - от нескольких месяцев до 15 и даже 20 лет. Средняя продолжительность жизни, по данным большинства авторов, равняется 4-6 годам, хотя многие больных живут до 8-10 лет и более. Больные погибают от разнообразных инфекционных, реже - от аутоиммунных осложнений, от анемии, кахексии, интоксикации, бластного криза в терминальном периоде заболевания.
При доброкачественном течении заболевания и проведении современной терапии большинство больных в течение длительного времени сохраняют и работоспособность.
В настоящее время общепринятая Международная классификация, позволяет определить прогноз заболевания.
Определение. Хронический лимфолейкоз (ХЛЛ) – моноклоновая опухоль из гемопоэтических клеток-предшественников, клеточные линии которой сохраняют способность дифференцироваться до морфологически зрелых, но функционально неполноценных лимфоцитов.
МКБ10: С91.1 – Хронический лимфоцитарный лейкоз.
Этиология. Этиологическим фактором ХЛЛ является вирусная инфекция. Запуск механизма болезни может происходить под воздействием сильного поля низкочастотных электромагнитных колебаний – проживание вблизи высоковольтных линий электропередач. Заболевают люди старшего возраста, чаще мужчины. У детей ХЛЛ не возникает.
Патогенез. Под воздействием этиологического и запускающего факторов возникает мутация в геноме гемопоэтической клетки предшественника с формированием опухолевого клона. Клеточные линии опухоли в процессе пролиферации способны дифференцироваться до морфологически зрелых, но функционально неполноценных В или Т лимфоцитов. В-тип опухоли возникает чаще (95% случаев), Т-тип – реже (5% случаев). Функциональный дефект В-лимфоцитов опухолевой линии заключается в неспособности трансформироваться в плазматические клетки.
Свидетельством генетических мутаций является появление лимфопоэтических клонов, имеющих хромосомные аберрации. Обнаруживаются трисомия 12-й хромосомы, делеция длинного плеча 13-й хромосомы, транслокации генов, нарушения их экспрессии.
Появление опухолевого клона функционально неполноценных лимфоцитов вызывает серьезные нарушения противовирусного и противоопухолевого иммунитета. По этой причине у больных ХЛЛ часто возникают и протекают в тяжелой форме инфекции герпевирусами, особенно II типа (опоясывающий лишай), опухолевые процессы с более злокачественным, чем ХЛЛ течением: лимфосаркома (синдром Рихтера), острый лейкоз и др.
Клиническая картина. Доброкачественная и прогрессирующая формы составляют классический ХЛЛ, клиническое течение которого проходит в три стадии: начальную, развернутую и терминальную.
Обычно увеличиваются шейные, надключичные, реже и во вторую очередь – подмышечные лимфоузлы. Другие группы вовлекаются в патологический процесс позже, на последующих стадиях ХЛЛ. Лимфоузлы различной величины. При пальпации они безболезненные, тестовато-эластичные, не спаяны между собой. Никогда не наблюдается изъязвлений или нагноений пораженных лимфоузлов.
Развернутая стадия ХЛЛ характеризуется прогрессирующим ухудшением самочувствия больных. Беспокоят выраженная общая слабость, похудение, субфебрильная лихорадка, зуд кожи, потливость по ночам. Появляются сердцебиения, одышка, кашель, боли в эпигастрии.
При осмотре обращает внимание выраженная лимфаденопатия. Выявляются одиночные или в виде пакетов увеличенные подчелюстные, шейные, подмышечные, паховые лимфоузлы, достигающие размеров куриного яйца. Пакеты лимфоузлов могут приводит к утолщению, нарушениям привычной формы шеи. При пальпации увеличенные лимфоузлы безболезненные, подвижные, тестовато-эластичные.
Обостряются кожные проявления псориаза, экземы, нейродермита, опоясывающего лишая. Могут возникнуть крапивница, разнообразные грибковые поражения.
Для Т-клеточного ХЛЛ характерны специфические лейкозные инфильтраты в глубоких слоях кожи с очаговым или диффузным поражением сосочкового и подсосочкового слоев.
У всех больных увеличивается селезенка. При спленомегалической форме ХЛЛ увеличение селезенки особенно значительное. Пальпаторно ее поверхность гладкая, плотная. Спленомегалии часто сопутствуют инфаркты селезенки, периспленит, проявляющимися интенсивной болью в левом подреберье, лихорадкой, шумом трения брюшины.
Гепатомегалия может быть выраженной умеренно. Край печени закругленный, плотный, пальпация безболезненная или слабо чувствительная.
Увеличенные медиастинальные лимфоузлы способны сдавливать органы средостения. Нередко возникают лейкемическая инфильтрация легких, плеврит. Клинически все это проявляется кашлем, одышкой, симптомами компрессии верхней полой вены с набуханием шейных вен, одутловатостью лица.
Выраженное увеличение внутрибрюшных лимфоузлов может являться причиной серьезных нарушений функции органов пищеварения, вплоть до механической желтухи, острого панкреатита, кишечной непроходимости. Лейкемическая инфильтрация стенки тонкой кишки проявляется поносами.
Терминальная стадия ХЛЛ наступает в связи с резким, трудно контролируемым утяжелением состояния больных. Для этой стадии характерно выраженное истощение, высокая лихорадка, часто обусловленные сопутствующими тяжелой пневмонией, обострившимся туберкулезом, генерализованной формой герпетической инфекции с поражением кожи, слизистых, дыхательных путей, желудочно-кишечного тракта, мочевыводящей системы.
Больные бледно-цианотичные, с одутловатым лицом, иктеричными склерами за счет анемии, холестаза, сердечной и легочной недостаточности, вызванной лейкозной инфильтрацией костного мозга, сердца, легких, сдавления лимфоузлами магистральных сосудов, инфильтративными изменениями в печени, компрессией желчных протоков. Возникает тромбоцитопеническая пурпура с многочисленными разноцветными кровоподтеками на коже.
Инфильтрация лейкозными клетками головного мозга проявляется мучительной головной болью, менингеальными симптомами, параплегиями.
Для терминальной стадии характерно особенно значительное увеличение всех групп лимфатических узлов, выраженная гепатомегалия. Гигантская селезенка, каменистая плотность быстро увеличивающихся лимфоузлов, прогрессирующие анемия, тромбоцитопеническая пурпура могут свидетельствовать о трансформации ХЛЛ в лимфосаркому (синдром Рихтера) или острый лейкоз.
Лейкозная инфильтрация почек приводит к быстро прогрессирующей почечной недостаточности. Это одна из причин гибели больных ХЛЛ.
Диагностика.
В начальную стадию заболевания:
Общий анализ крови: лейкоцитоз 10-50*10 9 /л за счет абсолютного лимфоцитоза. Остальные показатели нормальные.
В развернутую стадию заболевания:
Общий анализ крови: снижено содержание эритроцитов, гемоглобина, цветной показатель около единицы. Лейкоцитоз 50-200*10 9 /л за счет абсолютного лимфоцитоза. В мазке выявляются клетки Риделя (лимфоциты с двудольчатым почковидным ядром), тени Гумпрехта (фрагменты мембраны и ядер лимфоцитов, разорванных в процессе приготовления мазков). Тромбоцитопения.
Цитохимическое исследование: в опухолевых лимфоцитах повышенное содержание гликогена.
Стернальный пунктат: в клеточном составе костного мозга 50-60% лимфоидных клеток. Уменьшено содержание клеток гранулоцитарной, эритроцитарной, мегакариоцитарной линий.
Гистологическое исследование лимфоузла: в препарате признаки пролиферации лимфоидных клеток, большое количество морфологически зрелых форм. У больных с прогрессирующей формой ХЛЛ увеличено количество лимфобластов и пролимфоцитов.
Пункция селезенки: 90-98% клеточного состава пульпы являются лимфоцитами и пролимфоцитами.
Общий анализ мочи: протеинурия, микрогематурия.
Биохимический анализ крови: гипогаммаглобулинемия, умеренная гипербилирубинемия. Гиперурикемия не типична.
Иммунологический анализ: выявление принадлежности опухолевых лимфоцитов к В или Т популяциям.
Ультразвуковое исследование: объективизация спленомегалии, гепатомегалии, увеличенных лимфоузлов в брюшной полости. При саркоматозном поражении лимфоузлов их структура становится однородной, акустически прозрачной (гипоэхогенной).
В терминальную стадию заболевания:
Общий анализ крови: отклонения сходны с выявляемыми в развернутую стадию. В некоторых случаях лейкоцитоз может трансформироваться в лейкопению. Отмечается более глубокое уменьшение количества эритроцитов, гемоглобина. У всех имеет место выраженная тромбоцитопения.
Стернальный пунктат: тотальная лимфоидная инфильтрация костного мозга. При трансформации ХЛЛ в острый лейкоз резко увеличивается количество бластных клеток.
Отклонения других параметров сходны с возникающими в развернутую стадию ХЛЛ, но более выраженные.
Особенности клинико-лабораторных проявлений селезеночной, опухолевой, костномозговой форм ХЛЛ.
Селезеночная (спленомегалическая) форма. Проявляется выраженной спленомегалией, отсутствием гиперплазии лимфоузлов. В крови относительно небольшой лимфоцитоз.
Опухолевидная форма. Развивается у молодых людей. Отличается большими размерами увеличенных лимфоузлов при относительно небольшом лимфоцитозе (не более 10-20*10 9 /л) в периферической крови. Редко бывают анемия, тромбоцитопения, агранулоцитоз. В отпечатках лимфоузлов обнаруживают больше чем при типичных формах ХЛЛ лимфобластов, лимфоидных элементов с базофилией цитоплазмы.
Костномозговая форма. Характеризуется отсутствием лимфаденопатии, гепатоспленомегалии. В связи с тотальной инфильтрацией костного мозга лимфоцитами имеет место глубокая панцитопения (анемия, агранулоцитоз, тромбоцитопения).
Особенности клинико-лабораторных проявлений нозологически самостоятельных волосатоклеточного и пролимфоцитарного лимфолейкоза.
Волосатоклеточный лимфолейкоз (С91.4 по МКБ 10). При этом заболевании увеличиваются селезенка и печень. Периферические лимфоузлы небольших размеров. Характерна лимфопения. Сублейкемические и лейкемические варианты течения болезни встречаются редко. В крови уменьшено содержание эритроцитов, гранулоцитов, моноцитов, тромбоцитов. В мазках встречаются крупные "волосатые" лимфоциты.
Пролимфоцитарный лимфолейкоз (С91.3 по МКБ 10). Возникает у стариков. Встречается редко. Проявляется гиперлейкоцитозом, выраженной спленомегалией. Редко сопровождается выраженным увеличением лимфоузлов. Опухолевый клон лимфоидных элементов в крови, костном мозге, лимфоузлах представлен иммунологически более зрелыми клетками, чем при классическом ХЛЛ.
Международные критерии диагноза классического ХЛЛ:
Абсолютное количество лимфоцитов в периферической крови больше10*10 9 /л; большинство клеток являются морфологически зрелыми лимфоцитами.
Количество лимфоцитов в препаратах костно-мозгового пунктата больше 30% всех ядросодержащих клеток.
Большинство лимфоцитов периферической крови имеет иммунологические маркеры, подтверждающие их принадлежность к В-клеточному клону лейкозных клеток (маркеры В-лимфоцитов).
Диагноз считается обоснованным, если имеют место все три упомянутых выше критерия.
Дифференциальный диагноз. Проводится в первую очередь с реактивным лимфоцитозом и лимфаденопатией при вирусных и бактериальных инфекциях, аутоиммунных заболеваниях.
Отличить ХЛЛ может помочь характерное для этого заболевания сочетание лимфоцитоза и лимфаденопатии с гипогаммаглобулинемией. Функционально дефектные лимфоциты опухолевого клона не могут трансформироваться в плазмоциты – основной источник иммуноглобулинов. Наоборот, при реактивных лимфоцитозах с увеличением лимфатических узлов возникает гиперплазия нормальных клеточных линий лимфопоэза, которые достигают конечной точки своей дифференциации – плазматической клетки, способной синтезировать иммуноглобулины. В отличие от ХЛЛ в этих случаях возникает реактивная плазматическая инфильтрация костного мозга, гипергаммаглобулинемия.
План обследования.
Общий анализ крови.
Общий анализ мочи.
Биохимический анализ крови: общий белок и фракции.
Иммунологический анализ: содержание иммуноглобулинов всех классов. Типирование лимфоцитов на принадлежность к В-клеточному или Т-клеточному клонам.
Стернальная пункция и/или трепанобиопсия крыла подвздошной кости.
Гистологическое исследование лимфатических узлов.
Ультразвуковое исследование органов брюшной полости.
Рентгенография грудной клетки.
Лечение. На ранних стадиях заболевания, особенно при доброкачественном течении ХЛЛ медикаментозная терапия не проводится.
Прогрессирующая лимфаденопатия, выраженная спленомегалия являются основанием для проведения химиотерапии одним из цитостатиков:
Лейкеран 0,002 - по 1 таблетке 2 раза в день с переходом на поддерживающее лечение – 2 таблетки (0,004) 1 раз в неделю.
Циклофосфан 0,2 – вводить внутривенно по 400 мг 1 раз в день с последующим переходом на поддерживающие дозы – 400 мг внутривенно 2 раза в неделю.
Преднизолон назначают по 10-15 мг внутрь ежедневно на фоне лечения цитостатиками.
При злокачественных быстропрогрессирующих формах ХЛЛ и в терминальную стадию заболевания показано применение программ двух- трех- и четырехкомпонентной полихимиотерапии. Двухкомпонентная программа состоит из циклофосфана и винкристина. В трехкомпонентную программу ЦПП введены циклофосфан, пафенцил и преднизолон. В трехкомпонентной программе М-2 применены винкристин, циклофосфан, алкеран и преднизолон.
При глубокой тромбоцитопении и анемии проводят курс лечения большими дозами преднизолона - по 60-120 мг в сутки с последующим постепенным уменьшением дозы.
Применяют локальную лучевое воздействие на область увеличенной селезенки и пакеты лимфоузлов в дозе 1-1,5 Гр однократно, суммарно - 7-10 Гр.
Цитоферез проводят с целью удаления избытка лимфоцитов из периферической крови. На курс лечения - 5-8 сеансов.
Спленэктомия показана при формировании гемолитической анемии, глубокой тромбоцитопении, неэффективности глюкокортикоидов и лучевой терапии, частых инфарктах селезенки.
Для лечения инфекционных осложнений ХЛЛ применяют антибиотики широкого спектра действия, противогрибковые средства, инъекции препаратов иммуноглобулина.
Прогноз. Средняя продолжительность жизни больных хроническим лимфолейкозом 2-20 лет. После начала химиотерапии больные живут 4-6 лет.
Читайте также: