
Гетерозиготная форма неполипозного рака толстой кишки
Наследственный неполипозный рак толстой кишки (ННПРТК, синдром Линча I/II, синдром Х) - наиболее распространенная форма наследственного колоректального рака (КРР). В основе - аутосомно-доминантная мутация в генах репарации ошибочно спаренных оснований ДНК: hMLH1 или hMSH2 (90% мутаций в семьях с ННПРТК), hMSH6 (7-10%), PMS1 и PMS2 (5%).
а) Эпидемиология наследственного неполипозного рака толстой кишки (ННПРТК):
• Случаи заболевания составляют 3-5% от всего колоректального рака (КРР). ННПРТК: аутосомно-доминантное заболевание с пенетрантностью гена -80% и ускоренной последовательностью аденома - рак (2-3 года).
• Пожизненный риск развития рака толстой кишки отмечается приблизительно в 80% случаев, рака эндометрия - в 40-60%, рака мочевыводящих путей - в 18-20%, рака яичников - в 9-12%. Риск развития метахронного колоректального рака составляет 45% (после сегментарной резекции, 10-15% - после колэктомии с илеоректальным анастомозом); 70% опухолей расположены проксимальнее селезеночного изгиба.
б) Симптомы. Развитие колоректального рака (КРР) (и сопутствующих опухолей) в молодом возрасте. Симптомы отсутствуют или не отличаются от симптомов спорадического колоректального рака (КРР).
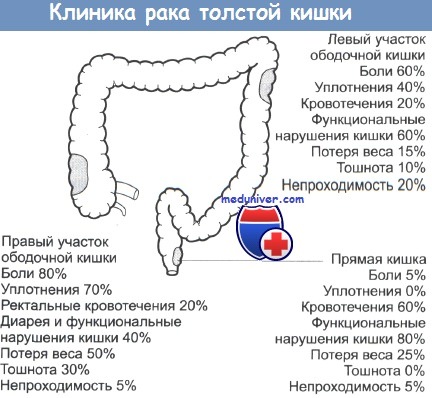
в) Дифференциальный диагноз наследственного неполипозного рака толстой кишки (ННПРТК):
• Другие варианты наследственного рака: САТК, МАП.
• Семейный КРР без идентификации мутантного гена.
г) Патоморфология:
• Макроскопическое исследование: ограниченное число полипов (часто плоских), в основном, в правых отделах (т.е. проксимальнее селезеночного изгиба).
• Микроскопическое исследование: обычно малодифференцированная аденокарцинома с медуллярным ростом, перстневидноклеточным и слизистым строением.
Микросателлитная нестабильность (MSI): 90-95% опухолей при ННПРТК MSI+, высокая частота MSI (изменения в двух и более из пяти панелей) по сравнению с 15-20% MSI+ при спорадическом КРР.
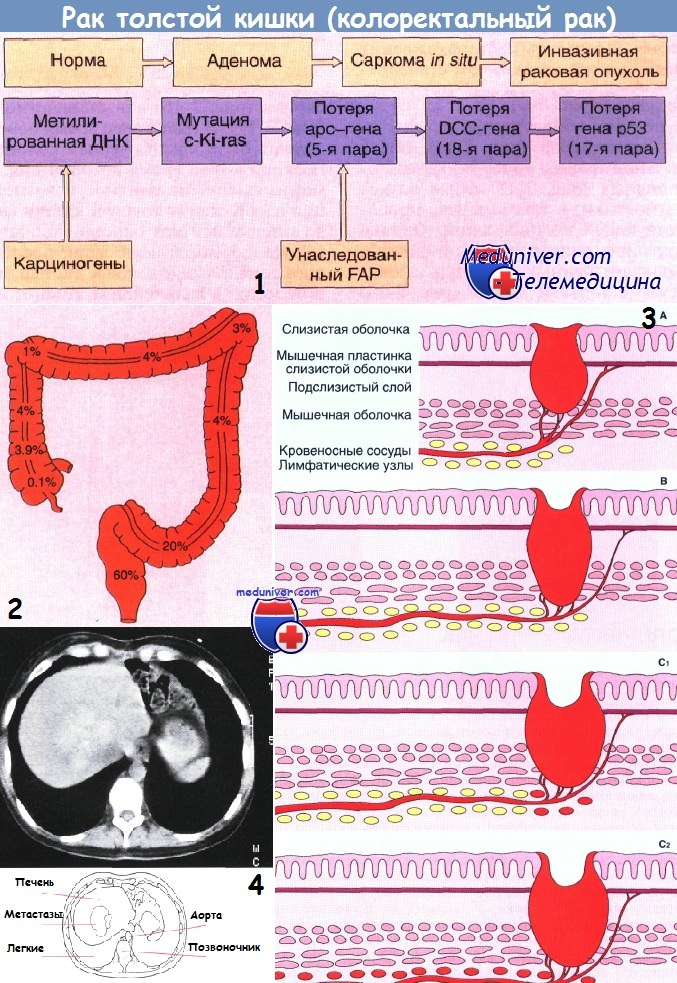
1 - Прогрессирование генетического дефекта, ведущее к развитию колоректального рака. Считают, что такая последовательность событий встречается часто, но необязательно включает все приведенные изменения и не всегда соответствует указанному порядку событий.
2 - Схема, иллюстрирующая частоту встречаемости рака в разных частях толстой кишки.
3 - Стадии развития рака толстой кишки по Дьюку (схема):
А — опухоль ограничена стенкой кишки;
В — прорастание мышечного слоя без вовлечения в процесс лимфатических узлов;
С1 — прорастание всех слоев стенки кишки с вовлечением ближайших лимфатических узлов;
С2 — то же, что и при стадии С1, плюс поражение отдаленных лимфатических узлов.
4 - Метастазы с центральным кальцинозом у больного с желтухой, вызванной диссеминированным колоректальным раком (без клинических симптомов поражения толстой кишки). Компьютерная томография.
д) Обследование при наследственном неполипозном раке толстой кишки (ННПРТК)
Необходимый минимальный стандарт:
• Выявление/лечение членов семьи с риском развития ННПРТК (у всех больных): запись в истории болезни о консультации и информировании пациента относительно данного заболевания (внимание: возможность судебного иска!)
Пациенты без симптомов/члены семьи при наличии семейного анамнеза:
• Семейный анамнез, генетическая консультация/тестирование (если не выполнено).
• Ежегодные колоноскопии, начиная с 25-летнего возраста (не позднее 10-15 лет с момента выявления рака у наиболее молодого члена семьи).
• Женщины: ежегодное исследование аспирата эндометрия.
• Скрининг других внекишечных опухолей: четкие рекомендации отсутствуют => в зависимости от семейных особенностей. При наличии рака, полипа с высокой степенью дисплазии или при увеличивающемся числе полипов => операция.
Больные колоректальным раком (КРР) моложе 50 лет:
• Семейный анамнез: Амстердамские критерии? Семья не соответствует критериям, если доказанность ННПРТК около 50%.
• Генетическое тестирование: критерии Бетезда?
• Идентификация/лечение членов семьи с риском развития ННПРТК: запись в истории болезни о консультации и информировании пациента относительно данного заболевания (внимание: возможность судебного иска!)
• Индивидуальная программа обследования для выявления КРР до операции.
Дополнительные исследования (необязательные) - такие же, как и при колоректальном раке (КРР).
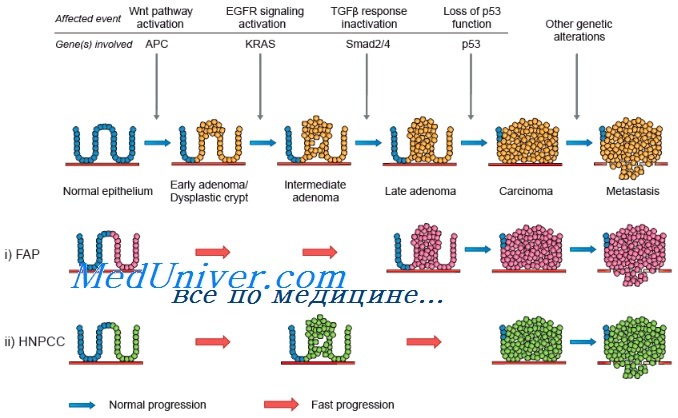
Генетика семейного рака кишечника
е) Классификация наследственного неполипозного рака толстой кишки (ННПРТК):
• Амстердамские критерии II: примерно в 50% семей, соответствующих критериям, выявляется ННПРТК.
• Критерии Бетезда МSI+, MSI-.
• Линч I: только колоректальный рак (КРР).
• Линч II: КРР и рак внекишечной локализации.
• Синдром Мюир - Торре: наследственный колоректальный рак (КРР) с опухолями сальных желез кожи.
• Синдром X: семейный колоректальный рак (КРР) неопределенного типа; выраженный семейный анамнез только колоректальный рак (КРР) (соответствует Амстердамским критериям, MSI- , нормальный ген ММР), более типична левосторонняя локализация, опухоли неслизеобразующие, немультифокальные, средний возраст выявления - 50 лет.
ж) Лечение без операции наследственного неполипозного рака толстой кишки (ННПРТК):
• Химиопрофилактика: роль не определена.
• Ежегодные колоноскопии и полипэктомии.
• В зависимости от стадии опухоли: адъювантная химиотерапия (рак толстой кишки), или (нео-) адъювантная химиолучевая терапия (рак прямой кишки).

а - Злокачественный полип нижних отделов толстой кишки. Ректороманоскопия гибким эндоскопом.
б - Колоноскопическая картина геморрагического рака восходящей ободочной кишки.
в - Рак правой половины толстой кишки.
г - Колоноскопическая картина рецидива рака в области анастомоза после удаления опухоли прямой кишки. 
з) Операция при наследственном неполипозном раке толстой кишки (ННПРТК)
Показания:
• Любой рак, прогрессирующая аденома (большой размер или дисплазия высокой степени) либо увеличение числа полипов, за исключением случаев множественных метастазов или абсолютных противопоказаний.
Хирургический подход:
• Субтотальная резекция ободочной кишки (лечебная + профилактическая): рекомендована при всех опухолях, расположенных проксимальнее сигмовидной кишки, за исключением случаев отказа больного или наличия противопоказаний к операции, связанной со значительным укорочением толстой кишки.
• Сегментарная колоректальная резекция: только с лечебной целью в соответствии с онкологическими принципами.
• Рак прямой кишки при ННПРТК: низкая передняя резекция (с/без неоадъювантной химиолучевой терапии), профилактическая проктоколэктомия обычно не рекомендуется из-за полной утраты функции прямой кишки.
• Женщины (в частности, при наличии семейного анамнеза рака матки или мутации в hMSH6 или в hMLHl и hMSH2): обсуждение вопроса о гистерэктомии/овариоэктомии (в пострепродуктивном периоде, при наступлении менопаузы или во время других абдоминальных вмешательств).
и) Результаты. Несмотря на худшие патоморфологические признаки при ННПРТК, влияние микросателлитной нестабильности (MSI) на ответ при химиотерапии по сравнению со спорадическим колоректальным раком (КРР) при сопоставлении в соответствии со стадией, все еще дискутабельно.
к) Наблюдение. Непрерывное ежегодное наблюдение/скрининг колоректального рака (КРР) и внекишечных опухолей => ежегодная полная колоноскопия, сигмоидоскопия (после операции), исследование аспирата эндометрия.
Рак толстой кишки в настоящее время — одно из самых распространенных онкологических заболеваний. Заболеваемость и смертность населения от рака продолжают неуклонно расти. Рак толстой кишки у мужчин занимает 3-е место, у женщин — 2-е. В 2009 г. в структуре заболеваемости злокачественными новообразованиями (ЗНО) в России КРР занял 2-е место. При этом показатель заболеваемости населения России раком ободочной кишки (РОК) составил 22,8 на 100 тыс. населения, а показатель заболеваемости ЗНО прямой кишки — 17,6. Максимальные уровни заболеваемости РОК зафиксированы в Санкт- Петербурге (37,7) и Москве (31,0). Очень эффективным есть лечение рака толстой кишки в Израиле.
Одним из основных факторов риска рака толстой кишки является пожилой возраст: риск возникновения повышается у лиц старше 55 лет и заметно возрастает после 70-75 лет.
В последнее время, несмотря на увеличение уровня заболеваемости в старших возрастных группах, все чаще РТК выявляется у молодых пациентов в возрасте до 45-50 лет, что наводит на мысль о наследственном характере данного заболевания.
Впервые мысль о предрасположенности некоторых семей к развитию рака в молодом возрасте и о возможности наследования онкологических заболеваний была высказана в 1895 г. В 40-50-х годах открываются редкие синдромы, предрасполагающие к развитию рака толстой кишки на фоне семейного полипоза: синдром Гарднера, Пейтца—Егерса, Тюрко и др., и устанавливается наследственный характер данного заболевания. В 70-90-х годах проводится анализ нескольких
тысяч родословных, на основании чего было показано, что подобные опухоли встречаются в 3 раза чаще, а частота рака других локализаций в изученных семьях не отличается от общепопуляционной.
Наиболее известными формами наследственного поражения толстой кишки являются семейный адено- матозный полипоз (САП) толстой кишки (familial adenomatous polyposys, FAP), частота развития рака на фоне которого составляет около 100 % и наследственный неполипозный рак толстой кишки, или синдром Линча.
На долю синдрома Линча приходится от 2 до 5 % всех случаев заболевания КРР .
Риск развития РТК у пациентов с ННПРТК составляет около 80 % .
Выделяют 2 основные формы ННПРТК :
- синдром Линча I — аутосомно-доминантное заболевание с изолированным поражением толстой кишки, преимущественно правых отделов, неполипоз- ной этиологии, возникающее в молодом возрасте и связанное с высоким риском развития опухолей толстой кишки;
- синдром Линча II — проявляется накоплением в семьях РТК, а также ЗНО других локализаций: рака эндометрия, яичников, молочной железы, желудка, тонкой кишки, поджелудочной железы, гепато-билиарного тракта и кожи (синдром Мюир-Торре) .
Клинические критерии ННПРТК
В 1991 г. международной группой исследователей впервые были согласованы клинические признаки ННПРТК, так называемые Амстердамские критерии. В мировой литературе они обозначены как Амстердамские критерии I (Amsterdam Criteria I) :
Молодой возраст возникновения заболевания (до 50 лет).
Наличие 3 или более родственников с морфологически верифицированным РТК.
Заболевание КРР более чем в 1 поколении.
Не меньше чем один из заболевших, должен быть родственником первой степени родства по отношению к остальным 2.
САП должен быть исключен.
В 1999 г. Амстердамские критерии I были расширены с включением злокачественных опухолей других локализаций. Они известны как Амстердамские критерии II (Amsterdam Criteria II) .
Новый дополнительный критерий к Амстердамским критериям I — наличие 3 или более родственников с ННПРТК-ассоциированными опухолями (РТК, рак эндометрия, тонкого кишечника, желудка, яичников, уретры, почечной лоханки, синдром Тюрко или синдром Мюир—Торре).
Рекомендации для проведения генетического консультирования при наличии клинических признаков — критерии Bethesda :
1. Рак, отвечающий Амстердамским критериям.
* Два наследственно-обусловленных рака, включая синхронные или метахронные злокачественные опухоли толстой кишки, или ассоциированные нео- плазии.
* Наличие КРР у пациента и кого-либо из родственников первой линии (либо ассоциированные неоплазии, либо аденоматозные полипы), при условии, что одна из злокачественных опухолей выявлена в возрасте до 40, а аденоматозный полип — в возрасте до 40 лет.
* КРР либо рак эндометрия, выявленный в возрасте до 45.
* Колоректальный аденоматозный полип, выявленный в возрасте до 40 лет.
Для более точной идентификации семей с ННПРТК в 2004 г. критерии Bethesda были обновлены (Revised Bethesda Guidelines) :
* КРР, возникший в возрасте до 50.
* Наличие синхронных, метахронных опухолей толстой кишки или ННПРТК-ассоциированных опухолей, независимо от возраста.
* КРР с повышенным уровнем микросателлитной нестабильности, диагностированный в возрасте до 60 лет.
* РТК, выявленный у 2 или больше родственников в любом возрасте.
В основе развития ННПРТК лежит наличие гер- миногенных мутаций в одном из генов репарации ДНК (ММЛ-гены) . Функция данных генов заключается в поддержании точности репликации ДНК.
В семьях, где помимо рака толстой кишки прослеживается накопление случаев заболевания раком тела матки, выявляются герминогенные мутации в гене MSH6. Предполагается, что МХН6-ассоциированная предрасположенность может реализовываться в атипичном течении и доброкачественных формах синдрома Линча. К тестированию других генов прибегают при отсутствии мутаций генов PMS1 и PMS2.
В случае наследования мутаций в генах MLH и MSH риск развития ЗНО толстой кишки увеличивается до 80 %. Для лиц мужского пола данные показатели еще выше. Риск развития КРР у лиц женского пола составляет 30 %, а риск развития рака эндометрия 40-60 %.
Нарушения в генах репарации ДНК приводят к увеличению частоты мутаций и связаны с фенотипом неустойчивости длины локуса микросателлита, т. е. они проявляют так называемую микросателлитную нестабильность .
Феномен микросателлитной нестабильности был открыт в 1993 г. М. Региса. Микросателлитная нестабильность определяется более чем в 90 % случаев ННПРТК и только в 10-15 % при спорадических формах КРР .
Именно поэтому первым молекулярным тестом для диагностики синдрома Линча является тест на микросателлитную нестабильность. При наличии положительного результата теста данные случаи подвергаются детальному генетическому тестированию на герминальные мутации в генах репарации ДНК.
Для ННПРТК характерен тот факт, что мутации в генах репарации ДНК передаются из поколения в поколение, т. е. от родителей к детям. У такого организма все клетки будут содержать как минимум одно онкологически значимое повреждение. Это приводит к ускорению канцерогенеза на одно звено .
Опухоли, ассоциируемые с мутациями MSH и MLH, характеризуются низкой степенью дифференцировки, по гистологическому строению чаще представлены слизистыми аденокарциномами и перстневидно-клеточным раком, для них характерно наличие лимфоцитарного инфильтрата, они редко метастазируют, лучше отвечают на лечение и имеют более благоприятный прогноз. Для лечения рака толстой кишки, лучше провести удаление толстой кишки в Израиле.
Диагностика ННПРТК представляет собой сложную задачу и складывается из тщательного анализа семейного анамнеза с последующим проведением генетического тестирования. Генетическая диагностика базируется на выявлении терминальных мутаций в генах репарации ДНК, в основном это 4 гена: MLH1, MSH2, MSH6и PMS2 .
Однако в клинической практике это занимает достаточно много времени и является дорогостоящим исследованием, поэтому генетическое тестирование всего населения практически невозможно и нецелесообразно.
Отделение колопроктологии НИИ онкологии им. Н.Н. Петрова одним из первых в нашей стране начало заниматься проведением генетического консультирования с целью диагностики наследственных форм КРР . Была показана необходимость создания групп повышенного риска (на основании Амстердамских критериев), которые необходимо подвергать генетическому консультированию.
Кроме того, было доказано, что ни один из клинических критериев в отдельности не является абсолютным и направление на генетическую консультацию нужно только при наличии не менее 3 клинических факторов.
В случае подтверждения в результате генетического консультирования наследственной природы КРР в послеоперационном периоде показано тщательное диспансерное наблюдение. Необходимо выделить 2 основные группы для диспансеризации. Прежде всего, это сами пациенты с синдромом Линча, подвергнутые хирургическому лечению, так как у них имеется повышенный риск развития метахронных новообразований и вторая группа — члены их семей. Учитывая тот факт, что возникновение ЗНО возможно в любом возрасте у лиц с наследственной предрасположенностью к КРР, клинический мониторинг за состоянием их здоровья должен осуществляться на протяжении всей жизни .
В настоящее время наиболее эффективным методом диагностики при наблюдении за лицами с высоким риском развития КРР и пациентами с ННПРТК является тотальная фиброколоноскопия.
Американская медицинская ассоциация рекомендует проводить фиброколоноскопию членам семей пациентов с синдромом Линча начиная с возраста 20—25 лет ежегодно или 1 раз в 2 года. В случае выявления герминогенных мутаций в гене MSH6 скрининг следует начинать с 30 лет . Пациентам с ННПРТК, подвергнутым хирургическому лечению, необходимо также ежегодно выполнять фиброколоноскопию.
Помимо КРР при синдроме Линча наиболее часто являются ЗНО женской репродуктивной системы: рак эндометрия (40—60 % женщин с герминогенными мутациями), рак яичников (12—15 %) , у женщин программа наблюдения должна быть дополнена трансвагинальным ультразвуковым исследованием малого таза, определением онкомаркера СА-125. Данная диагностическая программа наблюдения должна осуществляться ежегодно у пациенток с ННПРТК, которым было произведено хирургическое лечение, и у их родственниц начиная с возраста 30—35 лет.
Исследования по проблеме диагностики наследственных форм КРР на базе нашего отделения продолжаются. В частности, до настоящего времени нет работ, описывающих герминальные мутации, ответственные за предрасположенность к наследственному раку толстой кишки, встречающиеся в популяции Российской Федерации. Поиск устойчивых герминальных мутаций в популяции жителей России, детерминирующих предрасположенность к наследственному РТК, создание диагностической панели, возможность внедрения генетической диагностики в программу скрининга РТК, на наш взгляд, является перспективным направлением исследований на сегодняшний день. При поражении опухолью желудка проводится операция гастрэктомия - полное удаление желудка.
Наследственная неполипозная колоректальная карцинома – это наследственно обусловленная форма колоректального рака. Передается по аутосомно-доминантному типу. Чаще поражает проксимальные отделы толстого кишечника. Возможно одновременное или практически одновременное развитие нескольких злокачественных опухолей. Проявляется болями, нарушениями стула и явлениями кишечной непроходимости. Диагноз устанавливается на основании семейного анамнеза, клинической симптоматики и данных дополнительных исследований (колоноскопии, биопсии, генетических тестов). Лечение оперативное - радикальное или паллиативное.
МКБ-10
- Причины развития ННКРК
- Симптомы ННКРК
- Диагностика
- Лечение ННКРК
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Наследственная неполипозная колоректальная карцинома (ННКРК) – злокачественная опухоль толстой кишки, обусловленная наследственной мутацией. Составляет от 3 до 8% случаев колоректального рака. Относится к наследственным формам рака. Риск развития болезни в течение жизни при установленной мутации – около 80%. Злокачественная опухоль толстого кишечника нередко возникает в возрасте до 50 лет, то есть, на 10-15 лет раньше, чем в среднем по популяции. У пациентов с наследственной неполипозной колоректальной карциномой также отмечается повышенная вероятность развития рака яичников, рака эндометрия, рака тонкого кишечника, рака желудка, рака почек и рака поджелудочной железы. Суммарный риск развития злокачественных опухолей в течение жизни составляет более 90%. Лечение осуществляют специалисты в сфере клинической онкологии, проктологии и абдоминальной хирургии.
Причины развития ННКРК
Наличие подобных мутаций подтверждается при проведении генетических исследований. Вместе с тем, существуют клинические критерии, позволяющие обоснованно заподозрить наличие наследственной неполипозной колоректальной карциномы. Вероятность развития наследственного колоректального рака при выявлении данных критериев может варьировать из-за различия генетических поломок и разного уровня нестабильности клеток. К числу критериев относят:
- Наличие трех и более близких родственников с колоректальным раком, подтвержденным в ходе гистологического исследования. Один из трех родственников должен иметь первую степень родства с двумя другими. Наряду с колоректальным раком, учитывается наличие рака почки, тонкого кишечника и других органов, поражающихся при ННКРК.
- Развитие заболевания у представителей двух поколений.
- Хотя бы одно из новообразований было обнаружено у больного, не достигшего возраста 50 лет.
Для более точного прогнозирования специалисты-онкологи выделяют три группы пациентов с учетом склонности к развитию наследственной неполипозной колоректальной карциномы: группу низкого, среднего и высокого риска. В группу низкого риска входит большинство представителей популяции. У членов этой группы рак толстого кишечника в семейном анамнезе отсутствует либо выявляется у родственников не первой степени родства или у родственников первой степени родства в возрасте старше 45 лет. В группе среднего риска находятся больные, имеющие одного родственника первой степени родства, заболевшего колоректальным раком в возрасте младше 45 лет, либо двух родственников первой степени родства, заболевших в любом возрасте.
Группа высокого риска включает в себя членов семьи пациента с установленной наследственной неполипозной колоректальной карциномой и различными полипозными синдромами. Кроме того, в эту группу входят больные, у которых в семейном анамнезе выявляется три и более случаев рака толстого кишечника в пределах материнской или отцовской линии (у родителя, дедушки, бабушки, тети, дяди, сестры, брата, племянника и племянницы) либо два и более случаев рака толстого кишечника в пределах одной линии при условии, что хотя бы у одного родственника рак был множественным либо был диагностирован в возрасте до 45 лет.
Вероятность появления детей, страдающих наследственной неполипозной колоректальной карциномой при подтвержденном заболевании у одного из родителей, составляет 50%. Генетические исследования в период внутриутробного развития ребенка возможны, но на практике такую диагностику осуществляют очень редко из-за ее низкой практической значимости. Имеющаяся мутация может никак не проявиться в течение жизни (неполная пенетрантность гена) либо проявляться с различной степенью тяжести и в разном возрасте (вариабельная экспрессивность гена), поэтому оценить последствия наличия такого гена для ребенка заранее невозможно.
Симптомы ННКРК
Клиническая симптоматика соответствует классическому колоректальному раку. Отличительными особенностями являются более молодой возраст пациентов и преимущественное поражение проксимальных отделов толстого кишечника. Из-за высокого расположения наследственной неполипозной колоректальной карциномы кровь в кале визуально обычно не определяется, при значительной кровопотере возможен темный стул, при повторяющихся кровотечениях – анемия, проявляющаяся слабостью, головокружениями, бледностью кожных покровов и изменениями в анализе периферической крови.
При наследственной неполипозной колоректальной карциноме могут возникать явления кишечной непроходимости. Вначале кишечная непроходимость частичная, проявляется метеоризмом, задержкой стула и резкими схваткообразными болями, повторяющимися через 10-15 минут. При дальнейшем росте опухоли могут наблюдаться состояния, требующие экстренного хирургического вмешательства. Карциномы достаточно большого размера удается выявить при пальпации живота. При прогрессировании заболевания обнаруживаются общие признаки онкологического поражения: снижение аппетита, потеря веса вплоть до кахексии, слабость, гипертермия и т. д.
Диагностика
При постановке диагноза учитывается семейный анамнез, клинические проявления и данные дополнительных исследований. Больных направляют на колоноскопию, в процессе исследования осуществляют эндоскопическую биопсию опухолевой ткани. При подозрении на наличие наследственной неполипозной колоректальной карциномы наряду со стандартным морфологическим изучением материала выполняют исследование на микросателлитную изменчивость. При высоком уровне микросателлитной изменчивости проводят специальные генетические тесты.
Больным с наследственной неполипозной колоректальной карциномой назначают анализ кала на скрытую кровь, анализ периферической крови для выявления анемии и биохимический анализ крови для оценки функций печени. Для обнаружения метастазов и первичных опухолей другой локализации, возникающих при данной патологии, больных направляют на комплексное обследование, которое включает в себя гастроскопию, УЗИ органов малого таза, цитологическое исследование аспирата эндометрия, УЗИ брюшной полости, УЗИ забрюшинного пространства, рентгенографию грудной клетки, сцинтиграфию всего скелета и другие исследования.
Лечение ННКРК
Лечение хирургическое. С учетом высокого риска развития новых опухолей обычно осуществляют субтотальное удаление толстого кишечника с созданием илеоректального анастомоза. При вовлечении лимфатических узлов выполняют лимфаденэктомию. При наличии отдаленных метастазов проводят паллиативные оперативные вмешательства, направленные на устранение явлений кишечной непроходимости. Прогноз более благоприятный по сравнению с ненаследственными формами колоректального рака.
Прогноз и профилактика
Профилактика развития наследственной неполипозной колоректальной карциномы включает в себя регулярные обследования. Ближайшим родственникам пациентов с уже диагностированной ННКРК рекомендуют раз в два года проводить колоноскопию после 20 лет и раз в год – после 40 лет. Кроме того, им следует регулярно сдавать анализы мочи, а при появлении патологических изменений проходить обследование для исключения опухолей почек и мочеточников. Женщинам необходимо ежегодно обследоваться у гинеколога для исключения рака яичников и рака эндометрия. В некоторых случаях показана профилактическая пангистерэктомия. Дальним родственникам больных наследственной неполипозной колоректальной карциномой советуют провести генетическое исследование.








НАСЛЕДСТВЕННЫЙ НЕПОЛИПОЗНЫЙ РАК КИШЕЧНИКА
Онкология кишечника занимает 3 место в мире по сложности диагностики и лечения. Наследственный неполипозный рак кишечника(ННРК)является одним из наиболее часто встречающихся новообразований у человека. По крайней мере, у 50% индивидуумов в западных популяциях к 70 годам развиваются колоректальные опухоли, и приблизительно у 10% из них, в конечном счете, развивается колоректальный рак. Наследственный неполипозный рак кишечника встречается с частотой 2-5 на 1000, что составляет приблизительно 3-8% колоректального рака.
Цель: изучить проблему наследственно неполипозного рака кишечника
Изучить причины возникновения ННРК. Описать риск наследования, клинические признаки. Методы диагностики. Особенности ННРК. Первые признаки.
Наследственный неполипозный рак кишечника (ННРК) (MIM №120435) — генетически разнородный аутосомно-доминантный синдром предрасположенности к раку, часто вызывается мутациями в генах репарации неспаренных оснований ДНК. Наследственный неполипозный рак кишечника (ННРК) встречается с частотой 2-5 на 1000, что составляет приблизительно 3-8% колоректального рака.
Патогенез наследственного неполипозного рака кишечника.
При большинстве колоректальных опухолей, включая САП, кариотип опухоли прогрессивно становится все более анеуплоидным. Приблизительно 13-15% колоректальных опухолей не имеет такой хромосомной нестабильности, но имеет инсерцию или делецию в повторяющихся последовательностях (микросателлитная нестабильность). Микросателлитную нестабильность обнаруживают в 85-90% опухолей при наследственном неполипозном раке кишечника. В соответствии с этим наблюдением, приблизительно 70% семей наследственным неполипозным раком кишечника с карциномами, имеющими микросателлитную нестабильность, наследуют мутации в одном из шести генов репарации неспаренных оснований ДНК: MSH2, MSH6, MLH1, MLH3, PMS1 или PMS2. Репарация ДНК в неспаренных основаниях уменьшает ошибки репликации в 1000 раз. Ошибки синтеза ДНК вызывают неправильное спаривание нитей и деформируют двойную спираль ДНК. Комплекс белков репарации организует разные ферменты для восстановления повреждений. Используя вырезание длинных участков, этот комплекс удаляет ошибочный фрагмент вновь синтезированной нити ДНК и затем вновь синтезирует его. Чтобы вызывать микросателлитную нестабильность, должны потерять свою функцию оба аллеля гена репарации ДНК. Высокая частота соматической утраты функции второго аллеля приводит к тому, что наследственный неполипозный рак кишечника наследуется как аутосомно-доминантная болезнь приблизительно с 80% пенетрантностью. Соматическая утрата функции может происходить за счет потери гетерозиготности, внутригенных мутаций или гиперметилирования. При наследственном неполипозном раке кишечника количество мутантных микросателлитных локусов нарастает по мере перехода из аденомы в карциному. Инактивация генов, содержащих микросателлитные последовательности, может играть ключевую роль в развитии рака. Например, микросателлитная нестабильность порождает мутации типа сдвига рамки в гене рецептора фактора роста II (TGFBR2). Мутации в гене TGFBR2 приводят к утрате экспрессии белка TGF(3RII, и, поскольку система TGFP тормозит рост эпителиальных клеток толстого кишечника, переходу к неуправляемому росту. В поддержание концепции о роли гена TGFBR2 при наследственном неполипозном раке кишечника описана одна семья без мутаций в генах репарации ДНК, но с наследуемой мутацией в гене TGFBR2. Мутации гена TGFBR2 встречаются на ранних этапах наследственного неполипозного рака кишечника и могут содействовать росту аденом.
Фенотип и развитие наследственного неполипозного рака кишечника.
Хотя у больных ННРК число полипов аналогично общей популяции, они развиваются в более молодом возрасте. Средний возраст установки диагноза колоректальной аденокарциномы меньше 50 лет, т.е. на 10-15 лет раньше, чем в общей популяции. Больные наследственным неполипозным раком кишечника (ННРК) и известной унаследованной мутацией имеют 80% риск развития колоректальной опухоли в течение всей жизни. От 60 до 70% аденом и карцином при наследственном неполипозном раке кишечника (ННРК) развивается между селезеночным углом и илеоцекальным соединением, в то время как большинство спорадических колоректальных опухолей (а также опухоли при САП) развиваются в нисходящем отделе толстой кишки и сигмовидной кишке. Карциномы при ННРК реже имеют хромосомную нестабильность и ведут себя менее активно, чем спорадический рак кишечника; спорадические опухоли и карциномы при САП часто могут быть анеуплоидными и более энергичными. По этой причине больные наследственным неполипозным раком кишечника (ННРК) имеют лучший прогноз (с учетом стадии и возраста), чем больные с САП или колоректальными опухолями с хромосомной нестабильностью. Помимо колоректального рака, при наследственном неполипозном раке кишечника (ННРК) возможны и другие опухоли, включая рак желудка, тонкого кишечника, поджелудочной железы, почек, эндометрия и яичников; опухоли легкого и молочной железы с наследственным неполипозным раком кишечника (ННРК) не связаны. Пациенты с наследственным неполипозным раком кишечника (ННРК) и известной унаследованной мутацией в течение жизни имеют более 90% риска развития колоректального рака или одной из таких опухолей. Особенности фенотипических проявлений наследственного неполипозного рака кишечника: • Возраст начала: средний • Колоректальный рак • Многочисленные первичные опухоли
Особенности и опасность неполипозного рака толстой кишки
НРТК является наследственным заболеванием. Недуг может возникнуть по причине семейного рака (синдром Линча), или наследованием аденоматозных полипов.
В исследовании болезни очень важно иметь информацию о предшественниках раковых клеток, которые передались больному. Но, бывает так, что пациент не знает своих родственников, а тем более болезней, которым они подвергались. По этой причине – достоверно определить степень опасности и риск данной онкологии не представляется возможным.
Причины заболевания и кто в группе риска?
Риск развития заболевания у пациентов с наследственной предрасположенностью составляет 50%. Также, у таких людей, зачастую отсутствуют полипы в кишечнике, поэтому онкология развивается очень быстро. Существуют семьи, которые подвергаются опасности с поколения в поколение.
В большинстве случаев, ННРТК поражает людей с доминантным геном в возрасте до 60 лет. Для женщин риск составляет 91%, а для мужчин – 69%. Для синдрома Линча или ННРТК характерны следующие признаки:
Проявление заболевания у родственников в возрасте до 60 лет, в 2 – 3 поколениях.
Незрелый возраст проявления болезни у обоих родственников, в 2 поколениях.
Два случая любого вида онкологии у двух родственников в 2 – 3 поколениях. При этом, хоть один случай должен быть раком яичников, почечной лоханки, желудка, уретры или кишечника. В этом случае учитывается рак в возрасте только до 50 лет.
Случаи недуга у самых близких (отца и матери) в возрасте до 50 лет.
Диагностирование опухолей в 2 поколениях (до 50 лет).
Хотя-бы один родственник первой степени родства с диагнозом данной болезни.
При наличии генной мутации одного пациента, рекомендуется пройти обследование всем его родственникам. Таким образом, можно обнаружить опухоли на ранней стадии, что значительно увеличит шансы для лечения и полного выздоровления.
В самом начале своего развития, злокачественное образование в кишечнике растет практически без выраженных признаков. Только по мере его прогресса начинают проявляться такие симптомы:
Боли в животе. В зависимости от локализации опухоли, боль может быть разной: ноющей, давящей или в виде легких схваток.
Стабильный дискомфорт в желудке и кишечнике, а именно: чувство вздутия, частое газообразование и урчание.
Нерегулярный стул. Запоры и поносы могут чередоваться.
Возникновение рвотного рефлекса без причин, частые отрыжки и тошнота.
Чувство тяжести и переполнения желудка.
Помимо основных симптомов, ярким показателем образования являются кровяные сгустки в каловых массах. На цвет они бордово-коричневые, по причине сворачиваемости. Такой признак характерный для опухоли, расположенной в самом начале толстого кишечника. При обнаружении крови – следует незамедлительно обратиться к врачу!
Зрелая стадия ННРТК имеет разную симптоматику, в зависимости от места локализации образования. Клиническая картина условно разделяется на две стороны кишечника: правую и левую.
Опухоль в правой части сопровождается такими симптомами:
болевые ощущения, преимущественно, в правой стороне живота;
потеря сил, слабость всего организма;
наличие прощупывания образования.
Когда злокачественное образование развивается в правой стороне толстой кишки, у некоторых пациентов может повышаться температура. По статистике это 2 из 10 больных. Незначительная лихорадка может беспокоить человека долгое время, и многие считают это явление простудным синдромом. Однако, в такой ситуации, рекомендуется срочное обследование желудочно-кишечного тракта, так как повышение температуры – первый показатель правосторонней опухоли.
Симптоматика левостороннего ННРТК значительно отличается от недуга справа. Для рака такой локализации характерно возникновение нескольких мелких образований. Они являются препятствиями для равномерного распределения кала в кишечнике.
У пациентов данной категории наблюдаются продолжительные запоры. Задержка испражнения может длиться до 20 дней, так как плохо поддается медикаментозному лечению. Больной таким видом недуга может ощущать боли в животе, тяжесть и вздутие, что характерно для правостороннего рака. Однако, причиной этих симптомов является не опухоль, а
Необходимые анализы и обследования
Диагностика наследственной онкологии кишечника – довольно сложная задача. Для обследования необходимо детальное исследование семейного анамнеза и генетическое тестирование. На практике такой процесс является дорогостоящим и занимает много времени. По этой причине, обследовать все население невозможно и нецелесообразно.
При выявлении наследственности заболевания, врачи используют стандартные методы обследований, как для классического рака кишечника:
Врач прощупывает нижнюю область живота пациента. Методом пальпации специалист может определить наличие, размер образования.
Пациент должен сдать общий анализ крови на выявление анемии, анализ мочи, а также анализ кала на скрытую кровь и гельминтозы.
С помощью современных аппаратов специалист имеет возможность осмотреть сигмовидную и прямую кишки. Основные исследования – это томография, ректороманоскопия и колоноскопия (осмотр с помощью эндоскопа).
Для оценки состояния других органов, а также для диагностики возможных метастазов, проводиться рентгенография и УЗИ. А в последние годы самые точные показатели получают с помощью МРТ (магнитно – резонансной томографии).
Лечение наследственного неполипозного рака кишечника .
Диагноз наследственного неполипозного рака кишечника (ННРК) устанавливают на основе семейного анамнеза; пациенты не имеют различий в симптоматике. Минимальные критерии, позволяющие предположить наследственный неполипозный рак кишечника (ННРК) как причину случая колоректального рака или другой опухоли, — наличие наследственного неполипозного рака кишечника (ННРК) у трех членов семьи, по крайней мере, два из которых — первой степени родства, не менее чем в двух поколениях, причем развитие колоректальной опухоли, по крайней мере, у одного из больных должно произойти до 50 лет. У пациентов без семейного анамнеза, но с рано появившимся колорек-тальным раком показан ДНК-анализ клеток опухоли, позволяющий обнаружить микросателлитную нестабильность. Для диагностики применяют также иммуно-гистохимическое выявление белков MLH1 и MSH2. Раннее распознавание наследственного неполипозного рака кишечника (ННРК) необходимо для эффективного вмешательства; колоноскопический контроль проксимальных отделов толстой кишки, начатый в возрасте 25 лет, увеличивает ожидаемый срок жизни на 13,5 года, а профилактическое хирургическое удаление кишечника в возрасте 25 лет увеличивает ожидаемый срок жизни на 15,6 года. Биопсия эндометрия и абдоминальное УЗИ у женщин группы риска не показали эффективности в предупреждении рака матки, встречающегося при этом заболевании. В семьях с известными унаследованными мутациями идентификация мутаций в генах репарации ДНК помогает сфокусировать внимание на конкретных членах семьи, но если мутация неизвестна, это не отрицает потребности в частых осмотрах.
Риски наследования наследственного неполипозного рака кишечника.
Эмпирический общепопуляционный риск развития колоректального рака — 5-6%. Этот риск заметно изменяется в зависимости от семейного анамнеза. Относительный риск для пациентов, имеющих родственников первой степени с колоректальным раком, выше популя-ционного в 1,7 раза; он возрастает в 2,75 раза, если коло-ректальный рак наблюдали у двух и более родственников первой степени родства. Если опухоль у такого родственника развилась до 44 лет, относительный риск возрастает более чем в 5 раз. Обратите внимание, что пациент с унаследованной мутацией генов репарации неспаренных оснований ДНК имеет 50% риск родить ребенка, несущего ту же мутацию. Все дети, получившие такую мутацию, имеют приблизительно 90% риск развития опухолей в течение жизни, принимая пенетрантность наследственного неполипозного рака кишечника (ННРК) за 80%, плюс фоновый общепопуляционный риск опухолей кишечника и других опухолей, связанных с наследственным неполипозным раком кишечника (ННРК) (желудка, тонкой кишки, поджелудочной железы, почек, эндометрия, яичников). Пренатальная диагностика очень спорная и не входит в стандартные рекомендации, но теоретически возможна, если известна наследуемая мутация у родителя. Из-за неполной пенетрантности и варьирующей экспрессивности тяжесть и возраст появления рака кишечника и других опухолей предсказать невозможно.
Пример наследственного неполипозного рака кишечника.
40-летняя женщина, бухгалтер, мать троих детей, направлена в клинику онкогенетики лечащим врачом для консультации относительно ее семейного анамнеза опухолей. У ее отца, брата, племянника, племянницы, дяди по отцу и бабушки по отцу развился колоректальный рак. В анамнезе у пациентки не было терапевтических или хирургических проблем. Данные медицинского осмотра оказались нормальными. Генетик объяснил женщине, что ее семейный анамнез напоминает наследственный неполипозный рак кишечника, и наиболее эффективный способ определить генетическую причину рака в ее семье — молекулярный анализ живого больного члена семьи. После обсуждения с племянницей, единственной выжившей из больных членов семьи, женщина и племянница вернулись в клинику для обследования. Анализ архивного образца ткани опухоли, удаленной из кишечника, выявил микросателлитную нестабильность; последующее секвенирование ДНК из образца крови, полученного от племянницы, обнаружило наследуемую мутацию в гене MLH1. Сама пациентка не имела данной мутации; в связи с этим генетик заключил, что риск для нее самой и ее детей по развитию рака кишечника равен общепопуляционному. Однако оказалось, что у ее здорового брата мутация есть, и ему рекомендован постоянный ежегодный колоноскопический контроль.
Для своевременного обнаружения заболевания, а также для предотвращения неполипозного рака толстой кишки, необходимо следить за состоянием своего организма, а именно:
Периодически проходить осмотр у врача.
Незамедлительно лечить любые заболевания ЖКТ.
Соблюдать уровень витаминов А и С в организме.
Максимально правильно питаться.
Вести здоровый образ жизни и поддерживать физическое здоровье.
Наследственный неполипозный рак кишечника встречается с частотой 2-5 на 1000, что составляет приблизительно 3-8% колоректального рака. НРТК является наследственным заболеванием. Недуг может возникнуть по причине семейного рака (синдром Линча), или наследованием аденоматозных полипов. Основной причиной возникновения ННРТК является мутация гена, отвечающего за процессы в молекулах ДНК. Диагностика наследственной онкологии кишечника – довольно сложная задача. Для обследования необходимо детальное исследование семейного анамнеза и генетическое тестирование. На практике такой процесс является дорогостоящим и занимает много времени. По этой причине, обследовать все население невозможно и нецелесообразно. Раннее распознавание наследственного неполипозного рака кишечника (ННРК) необходимо для эффективного вмешательства
Список используемой литературы
2. Российский журнал Гастроэнтерологии ,Гепатологии ,Колопроктологии,2014
Читайте также: