
Генетика при острых лейкозах
Частое обнаружение изменений кариотипа в лейкозных клетках явилось предпосылками для дальнейшего прогресса в понимании патогенеза острого лейкоза, развития диагностических и прогностических моделей для различных вариантов острого лейкоза, оценки эффективности терапии и выявления раннего рецидива.
При остром лимфобластном лейкозе присутствие t(9;22), определяемое у 30% взрослых пациентов, t(4;ll), -7 или +8 (отмечаются нечасто) свидетельствуют о неблагоприятном прогнозе. Измененный кариотип наблюдается приблизительно у 80% больных острым миелобластным лейкозом. Выявление при ОМЛ t(15;17), t(8;21) и inv(16) прогностически благоприятно, в то время как +8, -5, del (5q), -7, del (7q), -20, +11, +13, inv (3) и вовлечение llq23 являются неблагоприятными признаками.
Определенные клинические варианты острого лейкоза, такие как М3, ассоциированы с t(15;17); миелоцитарный с созреванием — с t(8;21); острый миелобластный лейкоз с патологической костномозговой эозинофилией — с t(8;14); t(2;8); t(8;22) с inv (16) или del 16р; мегакариоцитарный — с t(l;22). Использование для лечения подофиллотоксинов или антрациклинов может привести к появлению транслокаций в течение 1-3 лет после завершения химиотерапии. После алкилирующих препаратов наиболее типично появление -5, -7 и комплексных хромосомных аномалий в течение 2-9 лет после завершения терапии.
Генетические особенности (цитогенетические и молекулярно-биологические), наряду с другими условиями (предшествующее лечение, миелодиспластический синдром (МДС) в анамнезе), существенно влияют на течение острого лейкоза, но далеко не всегда связаны с выделяемыми FAB-категориями. Недостатки FAB-классификации послужили причиной создания новой классификации, объединяющей генетические и клинические аспекты с морфологией, цитохимией и иммунофенотипом опухолей кроветворной системы. Работа по ее созданию была завершена в 1997 г. группой экспертов Европейской ассоциации гематопатологов и Общества гематопатологии.
Принятая ВОЗ-классификация более рациональна: каждая выделенная нозологическая форма имеет клиническое значение и может быть диагностирована патоморфологом.

Случаи с этими цитогенетическими нарушениями, но низким бластозом, относившиеся ранее к МДС, классифицируются как ОМЛ. Принято решение о включении в классификацию ВОЗ ОМЛ с мультилинейной дисплазией, ОМЛ с МДС в анамнезе и ОМЛ, связанного с проводившимся ранее лечением.
В 2001 г. в соответствии с решением комиссии ВОЗ сохранение FAB-терминологии и разделения острого лимфобластного лейкоза (ОЛЛ) на формы L1, 2 и 3 было признано нецелесообразным, так как прогностическая ценность морфологических вариантов L1 и L2, а также их корреляция с иммунофенотипом и цитогенетическими маркерами не очевидна, а форма L3 ОЛЛ является эквивалентом лимфомы Беркитта в фазе лейкемизации.
Изучение патогенеза опухолевых заболеваний системы крови привело к открытию одного из ведущих принципов онкогенеза в целом: причиной формирования злокачественного клона является нарушение функционирования нормальных генов. Как правило, это происходит в результате хромосомных аберраций, мутаций отдельных генов или блокирования нормальной регуляции функционирования генов в связи с эпигеномными (не связанными непосредственно с повреждением структуры генов) событиями.
Молекулярная генетика гемобластозов как отдельное научное направление стала развиваться в связи с успехами цитогенетики, которая, начавшись как дисциплина описательная, в дальнейшем позволила успешно идентифицировать гены, участвующие в хромосомных нарушениях.
В последние годы во всем мире продолжается активная исследовательская работа по идентификации генетических аномалий при гемобластозах и изучению их влияния на процессы жизнедеятельности клетки. Известно большое количество генетических нарушений, ведущих к развитию определенных видов опухолевых заболеваний крови. Детекция аномальных генов в настоящее время является обязательным условием для установления диагноза целого ряда гемобластозов.
Популярное ранее представление, что участие генов в транслокациях, нарушающих структуру их ДНК, само по себе придает им способность трансформировать клетку, сейчас кажется несколько упрощенным. Традиционно гены, участвующие в канцерогенезе классифицировались в одну из двух категорий: протоонкогены или опухолевые супрессоры. В настоящее время выделена третья категория: гены, поддерживающие целостность ДНК или обеспечивающие реконструкцию ДНК вследствие повреждений. Участвуя в аберрациях, эти гены не обладают способностью к трансформации, однако способствую накоплению трансформирующих мутаций.
Таким образом, эти три категории генов при любом виде повреждения прямым или непрямым способом ведут к малигнизации клетки.
Онкогены: исторически, гены, захваченные быстро трансформирующими ретровирусами из генома клетки называюся онкогенами, а их клеточные гомологи – протоонкогенами. В более широком смысле, онкогенами называются любые гены, образование доминантных мутаций в которых ведет к малигнизации клетки.
В настоящее время известно боле 30 ретровирусных онкогенов, имеющих клеточные аналоги. Лишь небольшая часть из них участвует в транслокациях, приводящих к развитию гемобластозов, но практически все задействованы в основных регуляторных путях. Гены, вовлеченные в транслокации, могут вызывать трансформацию клетки с помощью двух механизмов. Классическим примером первого из них является транслокация t(9;22), когда происходит формирование химерного онкогена bcr/abl, белок которого – тирозинкиназа – обладает непосредственным трансформирующим потенциалом.
Гены-супрессоры опухоли: Обнаружение хромосомных делеций, часто возникающих при определенных видах опухолей, привело к выявлению генов-супрессоров опухоли, потеря которых вызывает злокачественную трансформацию клетки. Подразумевается, что при потере одного гена одновременно должна происходить механическая или функциональная потеря такого же гена на второй хромосоме, для того, чтобы реализовался негативный эффект мутации.
Этот механизм часто задействован при солидных опухолях (например, RB1 при ретинобластоме, TP53 при опухолях толстого кишечника). Известно около 30 регионов, расположенных на 13 хромосомах, в которых регулярно происходят делеции или обнаруживается потеря гетерозиготности, ведущие к развитию онкогематологических заболеваний.
Гены, поддерживающие целостность ДНК. Мутации в этих генах не приводят непосредственно к развитию опухолей, однако, в результате возрастания генетической нестабильности, способствуют накоплению мутаций в клетке. Зачастую эти гены, относящиеся к микросателлитам (например, MLH1 и MSH2) мутируют в случаях солидных опухолей. Для гемобластозов такие виды мутаций нехарактерны.
Отражением другого вида генетической нестабильности являются множественные хромосомные аберрации у пациентов ОМЛ и МДС из группы высокого риска. Как правило, течение таких видов лейкемии сопровождается быстрым развитием резистентности к терапии или возникновением ранних рецидивов. Очевидно, наличие множественных хромосомных аберраций является отражением ранних событий в лейкемической клетке, скорее всего, повреждением механизма восстановления двухцепочечной ДНК. Известен ряд генов, которые, возможно, вовлечены в этот процесс (ATM, TP53, MRE11).
Генетические аномалии при острых миелоидных лейкозах
Основными молекулярными событиями, ведущими к формированию лейкемического клона при миелоидных лейкозах, являются либо возникновение специфических транслокаций, зачастую с вовлечением протоокогенов, либо мутации генов, участвующих в контроле пролиферации и дифференцировки миелоидной ткани. Для миелоидных опухолей наиболее характерными являются реципрокные транслокации, при которых происходит обмен генетическим материалом между различными хромосомами с образованием патологических хромосомных структур, самой известной из которых является Филадельфийская хромосома.
На молекулярном уровне в процессе такой транслокации образуется так называемый химерный ген, состоящий из активных участков (доменов) двух генов-участников перестройки и ведущий к экспрессии химерного белка, который способен, как правило, либо блокировать миелоидную дифференцировку, либо стимулировать бесконтрольную клеточную пролиферации за счет следующих событий:
- нарушение функционирования ядерных рецепторов (характерный пример – острый промиелоцитарный лейкоз);
- подавление транскрипции (считывания РНК с ДНК) за счет связывания ключевого белкового комплекса CBF (core binding factor), что характерно для острых лейкозов с транслокациями, вовлекающими 21 и 16 хромосомы (гены RUNX-1, бывший AML-1, и CBFB);
- подавление генов гомеобокса, или регуляторов клеточного развития (характерно для ОМЛ с транслокациями, в которых участвует ген MLL – mixed lineage leukemia-, расположенный на 11 хромосоме);
- бесконтрольная активация ферментов-тирозинкиназ (BCR-ABL позитивные миелоидные лейкозы, мутации гена FLT3);
Особо хотелось бы отметить важность мутаций генов, не вовлеченных в транслокации, но являющихся медиаторами процессов, описанных выше. К ним относятся частичная тандемная дупликация гена MLL в случае соматической мутации 11q23, внутренняя тандемная дупликация гена FLT3, ведущая к активации киназного каскада за счет маскировки под нормальный киназный рецептор, мутации генов СЕВРА и RUNX1 (AML1), подавляющих транскрипцию за счет связывания комплекса CBF, мутации гена NPМ, приводящие к блоку образования белков в рибосомах.
Значение химерного гена PML/RARA при остром промиелоцитарном лейкозе и его роль в блоке миелоидной дифференцировки
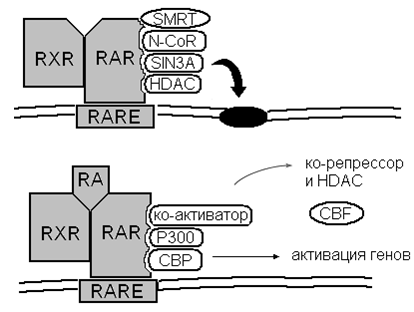
На рис. 3 изображена нормальная регуляция миелоидной дифференцировки в результате активации рецептора ретиноевой кислоты α (RARα). В неактивном виде рецептор образует подавляющий транскрипцию комплекс вместе с другим рецептором этого же семейства (RXR), ядерными помощниками – корепрессорами (SMRT, N-CoR, Sin3a) и белком гистондеацетилазой HDAC.
Этот комплекс соединяется со специфическим участком ДНК (RARE – retinoid acid responsive element), полностью блокируя считывание РНК. В нормальной ситуации рецептор связывается с ретиноевой кислотой (RA) и связь с корепрессорами разрушается. Далее, вместо корепрессоров, происходит присоединение коактиваторов (P300, CBP), что приводит к активации нормальной миелоидной дифференцировки.
При образовании транслокации t(15;17), ведущей к появлению химерного белка PML/RARA, возникает прочная связь рецептора с корепрессорами, не разрушающаяся в присутствии физиологических концентраций полностью транс-ретиноевой кислоты (ATRA). ДНК освобождается лишь в присутствии фармакологических концентраций ATRA, превышающих физиологическую в тысячи раз. Только это делает возможной нормальное функционирование белкового комплекса и дальнейшую миелоидную дифференцировку (10).
Тот же механизм работает и при транслокации t(5;17), вовлекающей гены NPM и RARA. Совершенно другая ситуация возникает при ОПЛ с транслокацией t(11;17) (PLZF/RARA) и t(11;17) STAT5b/RARA. Ген PLZF, так же как и STAT5b, имеет собственные зоны контакта с комплексом корепрессоров и добавление ретиноевой кислоты не приводит к освобождению зависимых генов для нормальной экспрессии. Сложность диагностики при остром промиелоцитарном лейкозе заключается в том, что морфологические и цитохимические методы не позволяют различать между собой разные виды характерных транслокаций.
Стандартная цитогенетика зачастую неэффективна, так как при ОПЛ клетки костного мозга в обычной краткосрочной культуре делятся плохо, и материала не хватает для подтверждения диагноза ОПЛ и дифференцирования транслокаций. Все вышеуказанные транслокации возможно определять с помощью флюоресцентной in situ гибридизации (FISH) или метода обратной транскриптазной реакции с проведением далее так называемой гнездной полимеразной цепной реакции.
Рис. 3. Нормальная регуляция миелоидной дифференцировки путем активации рецептора ретиноевой кислоты.
Таким образом, определение транслокаций при ОПЛ является крайне необходимым не только для установления правильного диагноза, но и для определения первичной резистентности к стандартной для ОПЛ терапии ATRA и, соответственно, к определению прогноза (благоприятного при t(15;17) и t(5;17) и неблагоприятного при всех остальных видах транслокаций).
Значение определения транслокаций, участвущих в репрессии комплекса CBF
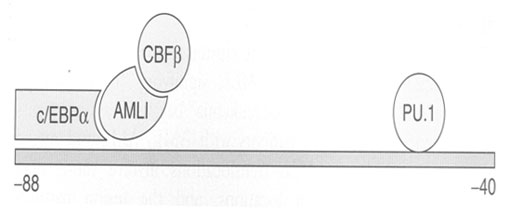
Образование нормального комплекса CBF, ведущего к активации гена макрофагального колониестимулирующего фактора (M-CSF), играет важнейшую роль в миелоидной дифференцировке и напрямую зависит от формирования связи между белками-продуктами генов c/EBPα, AML1(RUNX1) и CBFβ (рис 4). Поэтому, все транслокации, вовлекающие хотя бы один из перечисленных генов, такие как t(8;21) или inv 16 превращают указанный комплекс из активатора в репрессор, и таким образом блокируют миелоидную дифференцировку.
Блокада происходит с участием ядерных корепрессоров и гистондеацетилаз, тех же что были описаны в предыдущем разделе, касающемся лейкемогенеза при ОПЛ. Надо отметить, что любые мутации генов, участников комплекса CBF, таких как c/EBPα, RUNX1, даже при отсутствии цитогенетически определяемых нарушений ведут к формированию лейкемического фенотипа, то есть, нарушению дифференцировки и бесконтрольной пролиферации. Тем не менее, по современной классификации, обнаружение данных нарушений относят этот тип лейкоза в благоприятную группу (при условии проведения соответствующей терапии).
Рис. 4. Образование комплекса CBF
Обычно транслокации t(8;21) или inv 16 и др. довольно легко определяются при стандартном цитогенетическом исследовании. Так же, как и при ОПЛ, используется обратно-транскриптазная полимеразная цепная реакция. При определении мутаций генов c/EBPα, RUNX1 с наибольшей эффективностью может быть применена только ПЦР.
Если учесть, что данные аберрации суммарно встречаются примерно в 15-20% М2 без цитогенетически выявляемых нарушений, становится ясной необходимость выявления этих мутаций, тем более, что по последним данным, обнаружение таких аномалий без сочетания с другими цитогенетическими нарушениями относит этот тип лейкоза из группы промежуточного в группу благоприятного прогноза.
Определение транслокаций, ведущих к активации тирозинкиназ и киназных рецепторов.
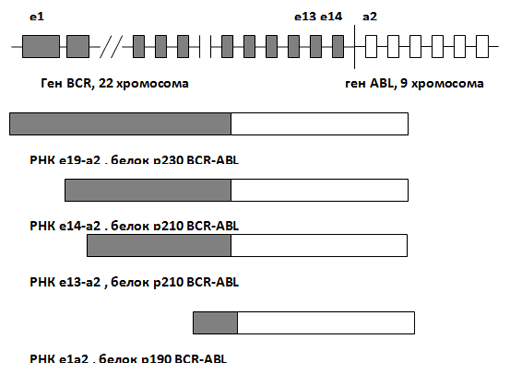
Самой известной транслокацией, приводящей к аномальной активации киназного каскада и неконтролируемой клеточной пролиферации, является Филадельфийская хромосома или t(9;22), ведущая к образованию химерного гена BCR/ABL (рис 5). В зависимости от локализации точки разрыва на 22 хромосоме образуется химерный онкоген различной длины (от 190 до 230 килодальтон).
Продукт этого гена (тирозинкиназа) ведет к фосфорилированию огромного количества протеинов (RAS, PI-3, Grb2, Crkl), участвующих в процессе сигнальной трансдукции – передаче сигнала с рецепторов клеточной мембраны ядерному генетическому материалу. Активация такого количества различных сигнальных путей ведет к независимой от ростовых факторов пролиферации, нарушению адгезии клеток к стромальному окружению и устойчивости к апоптозу – программированной клеточной гибели. При остром миелоидном лейкозе встречается как тип транслокации с точкой разрыва M-bcr, ведущий к образованию протеина размером 210 килодальтон (р210), так и m-bcr, с образованием протеина p190.
Рис. 5. Образование химерного гена BCR/ABL.
Другим вариантом самопроизвольной стимуляции киназного каскада является выявление мутации гена FLT3 (FMS-like tyrosine kinase receptor), кодирующего тирозин-киназный рецептор. Образование так называемой внутренней тандемной дупликации в структуре гена ведет к изменению третичной структуры рецептора таким образом, что он мимикрирует под обычный рецептор, уже активированный специфическим лигандом.
Это ведет к активации протеинкиназ ERK2 и AKT, приводит к неконтролируемой пролиферации и обычно ассоциируется с выраженным лейкоцитозом. Следует особо отметить, что выявление этого нарушения у пациентов с t(15;17) или у больных без явных нарушений кариотипа резко ухудшает прогноз и переводит этих пациентов в группу неблагоприятного течения ОМЛ.
По данным последних исследований, все виды нарушений, связанных с активацией киназных сигнальных путей являются независимым отрицательным прогностическим признаком.
Значение выявления транслокаций, ведущих к нарушению экспрессии генов гомеобокса
К генам гомеобокса относят целый ряд генов, кодирующих транскрипционные факторы, которые принимают участие в регуляции эмбрионального развития. Эти гены имеют высоко гомологичную структуру у большинства эукариот, от самых низших до наиболее высоко организованных. Наиболее значимым геном-регулятором экспрессии генов гомеобокса у млекопитающих является ген MLL (mixed lineage leukemia).
Партнерами этого гена являются гены-регуляторы транскрипции, поэтому любые мутации, вовлекающие MLL, ведут к подавлению транскрипции генов гомеобокса. Точный механизм лейкемогенеза в данном случае пока до конца не определен. MLL участвует в образовании более 25 различных транслокаций, которые (кроме t(4;11)) обычно характерны для ОМЛ М4/М5. Наиболее известным и распространенным нарушением является соматическая мутация 11q23, приводящая к образованию частичной внутренней тандемной дупликации, в которой участвуют экзоны со 2 по 6 или со 2 по 8. Подобное нарушение определяется в случаях трисомии 11 хромосомы и в 11-20% случаев ОМЛ без цитогенетических нарушений. Следует отметить, что любое нарушение структуры гена MLL однозначно определяет неблагоприятный вариант течения ОМЛ.
Мутации гена NPM
Ген нуклеофозмина (NPM-1) в последнее время привлекает все более пристальное внимание исследователей, ввиду частой выявляемости мутаций этого гена при ОМЛ с нормальным кариотипом (50%-60%). В норме продукт этого гена играет ключевую роль в образовании протеосом и, таким образом, опосредованно влияет на синтез белков в клетке. Ген NPM-1 участвует в ряде реципрокных транслокаций, характерных для анаплазированной Т-клеточной лимфомы t(2;5), редко встречающихся форм МДС с транслокацией t(3;5), и крайне редкого типа ОПЛ с t(5;17).
Для ОМЛ более характерны мутации NPM-1, наиболее частой из которых является дупликация тетрануклеотида TCTG в позиции 956-959 в 5 экзоне (75-80% случаев). Во всех случаях мутаций или транслокаций с участием NPM-1 происходит изменение нормального внутриядерного расположения белка на цитоплазматическое и нарушается его взаимодействие с генами – партнерами (ТР53 и ARF), что инактивирует их как опухолевые супрессоры. Тем не менее, обнаружение мутаций NPM-1, как правило, ассоциируется с благоприятным течением ОМЛ (за исключением случаев ассоциации с мутациями гена FLT-3). В настоящее время продолжается активное изучение этого гена с целью уточнения его роли в лейкемогенезе и возможного использования его как маркера при исследовании минимальной остаточной болезни.
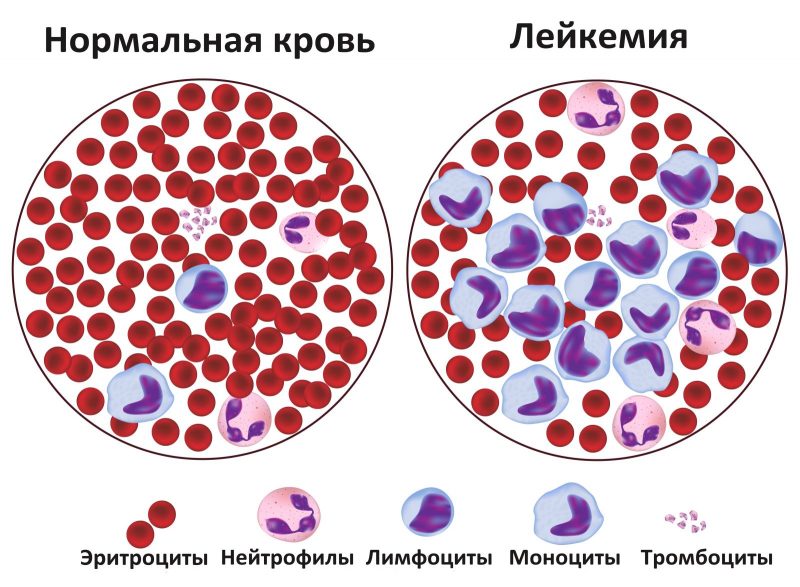
Кровь представляет собой жидкую ткань организма, которая состоит из красных и белых клеток. К первым относятся эритроциты и тромбоциты. Белые представлены гранулоцитами и агранулоцитами. Свое название они получили в результате микроскопического исследования: те клетки, в которых обнаружены гранулы, — гранулоциты. Соответственно, агранулоциты их не имеют. Каждая из фракций крови выполняет определенную функцию. При лейкозе нарушаются функции всех органов и систем человеческого организма.
В составе крови существует четкая пропорция соотношения красных клеток к белым. Ее изменение определяется как лейкоз, то есть разрастание клеток крови одного из ростков — белого или красного. Чаще всего опухолевым изменениям подвергаются белые ростки, поэтому лейкоз иначе называют лейкемией. Опухоль злокачественная, но не называется раком, потому что раковые клетки зарождаются только в эпителиальной ткани.
Причины развития лейкозов
Поражение ростков крови приводит к повреждениям зрелых клеток, которые имеют все признаки опухоли, следовательно, не выполняют полезных функций. Ростки называются мегалобластами, поэтому лейкозы имеют второе название — мегалобластозы. Их изменения происходят на генном уровне в результате необратимых мутаций. Этому способствуют следующие факторы:
- наследственная предрасположенность — чаще всего характерна для хронических лейкозов;
- радиационное поражение — одна из форм лучевой болезни;
- неблагоприятная экологическая обстановка — воздействие канцерогенных факторов;
- синдром иммунодефицита.
Опухоль зарождается в красном костном мозге. По мере развития лейкоза опухолевые клетки замещают здоровые ростки кроветворения. Попадая в общий кровоток, они метастазируют во все органы и ткани организма. Продолжая размножаться на новых местах локализации, клетки вновь циркулируют по кровеносной системе. В зависимости от того, на каком этапе кроветворения происходит генная мутация, лейкозы подразделяются на острые и хронические.
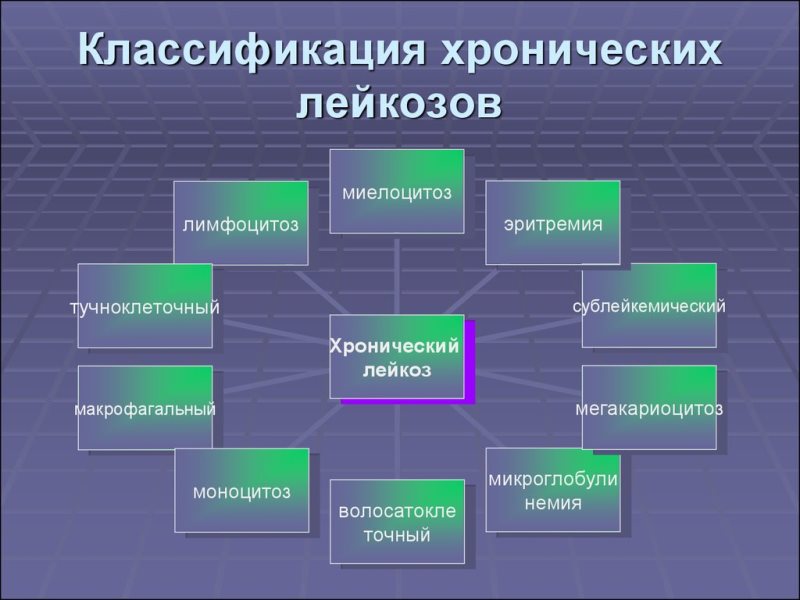
Классификация лейкозов
Классификация гемобластозов основана на морфологических признаках. Острый лейкоз развивается при поражении унипотентной, стволовой клетки крови. Названия хронических форм зависят от того, какие зрелые клетки крови поражены опухолью. Острая форма лейкоза никогда не станет хронической, которая, в свою очередь, по характеру течения ничем не отличается от острой.
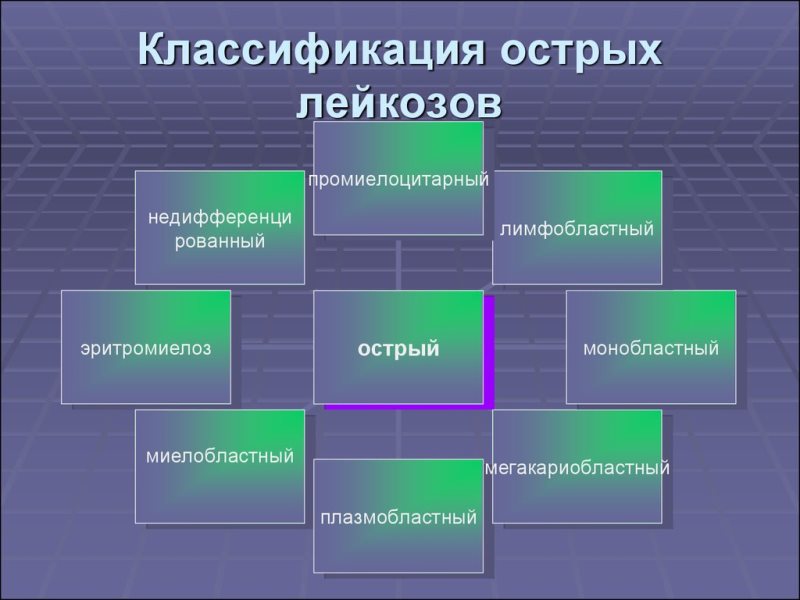
Формы острых лейкозов в зависимости от уровня поражения системы кроветворения:
- миелобластная — стволовые клетки;
- монобластная — моноциты (агранулоциты);
- лимфобластная — лимфоциты (гранулоциты);
- эритромиелобластная — предшественники эритроцитов;
- мегакариобластная — тромбоциты (мегакариоциты);
- недифференцированная — ростки всех клеток крови.
- миелоцитарные, нейтрофильные, эозинофильные, базофильные, миелоидные, тромбоцитемические, эритремические;
- лимфоцитарные — лимфолейкоз, парапротеинемические формы — миеломная и болезнь Сезари, макроглобулинемия Вальденстрема, болезнь Франклина;
- моноцитарные, миеломоноцитарные, гистиоцитоз X.
Симптомы лейкоза:
- боли в костях и суставах;
- кровоточивость;
- увеличение лимфатических узлов, печени, селезенки;
- состояние иммунодефицита — снижение сопротивляемости к инфекциям;
- менингит и энцефалит вследствие метастазов в головной мозг;
- общие явления интоксикации, лихорадка, быстрое похудение, слабость.
Диагностика лейкозов
Диагностические критерии определяются в лаборатории. В зависимости от того, какая именно клетка поражена, выставляется верный диагноз. Генетические исследования позволяют определить стадию и тяжесть опухолевого процесса. Обнаружение экспрессии определенных групп генов и филадельфийской хромосомы имеет значение для выбора препаратов для химиотерапии и своевременного решения о трансплантации красного костного мозга.
Лечение лейкоза
Независимо от формы заболевания, в первую очередь производится уничтожение всех опухолевых клеток — как в месте образования, так и в метастазах. При лейкозах применяется химиотерапия цитостатиками — препаратами, прекращающими рост опухоли. Подбираются средства, к которым чувствительны злокачественные клетки крови. После курса химиотерапии при острых лейкозах производится трансплантация красного костного мозга или проводится повторный курс лечения цитостатиками такой же длительностью и в такой же дозировке. Третий курс химиотерапии — профилактический. При хронических формах лейкоза применяется оперативное лечение и радиотерапия.








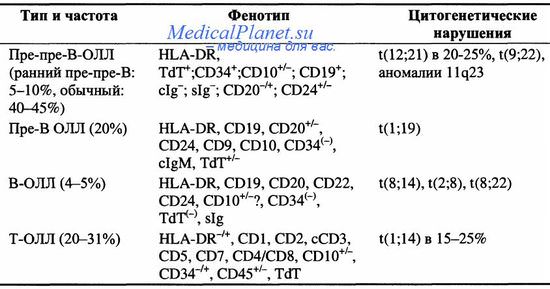
ЦИТОГЕНЕТИЧЕСКИЕ АНОМАЛИИ ПРИ ОСТРОМ ЛИМФОБЛАСТНОМ ЛЕЙКОЗЕ
Актуальность. В последнее время отмечается тенденция к росту злокачественной патологии, как взрослого населения, так и детей. В педиатрической онкологии острый лимфобластный лейкоз (ОЛЛ) является одним из самых распространённых заболеваний гемопоэтической ткани и составляет до 30% всех опухолей, до 75% всех гемобластозов [1]. Стандартный цитогенетический метод исследования является необходимым в проведении диагностики у пациента с подозрением на ОЛ. Выявление количественных и структурных изменений хромосом позволяет установить прогностическое значение данного заболевания.
Цель. Изучить структуру острых лейкозов, выявленных с помощью цитогенетического исследования и прогноз часто встречающихся цитогенетических нарушений при ОЛЛ.
Цитогенетический метод заключается в микрокопировании структуры хромосом и их количества на основании исследования клеток периферической крови . Под микроскопом могут обнаруживаться лишь хромосомные и геномные мутации. Данный метод исследования стали широко применять в генетике с 1956 года, когда шведские ученые Дж. Тийо и А. Леван предложили новый способ изучения хромосом, установив верный кариотип. Впервые прямая связь между конкретной хромосомной перестройкой и определенным типом злокачественно заболевания была установлена в 1960 году Новеллом и Хунгерфордом. Современный этап в применении цитогенетического метода связан с разработкой дифференциального окрашивания хромосом Т. Касперсоном в 1969 г. Это позволило расширить возможности цитогенетического анализа и точно идентифицировать хромосомы по характеру распределения в них окрашиваемых сегментов. Сейчас известно множество примеров хромосомных перестроек, которые определяют склонность к развитию онкологических заболеваний или являются самой причиной.
Стандартный цитогенетический метод входит в список обязательных диагностических процедур, проводимых у больных с подозрением на ОЛ. Кариотип пациента исследуется по клеткам периферической крови. Для того, чтобы считать кариотип достоверным, необходимо провести исследование не менее 20 метафаз. К основным типам генетических аномалий при ОЛЛ относятся количественные аномалии, т.е. нарушения плоидности, и структурные аномалии, к которым относятся транслокации, инверсии, делеции, дупликации и точковые мутации. К количественным хромосомным аномалиям относятся гипер- и гипоплоидия. Гиперплоидия характеризуется приобретением дополнительных хромосом с их увеличением больше 46 в одной клетке. Она обнаруживается в 5-15 % случаев ОЛЛ у взрослых, что снижает вероятность благоприятного прогноза, нежели при ОЛЛ у детей, когда гиперплоидность обнаруживается приблизительно в трети случаев. Гипоплоидия, наоборот, является следствием снижения количества хромосом меньше 46. Она обнаруживается в 2-8% случаев ОЛЛ и ассоциируется с неблагоприятным исходом [1]. Качественные (структурные) хромосомные аномалии при ОЛЛ обнаруживаются чаще. Хромосомные транслокации являются, как правило, первичными генетическими событиями, формирующимися на ранних этапах лейкемогенеза в то время как точковые мутации, делеции чаще оказываются вторичными аномалиями, приобретенными в результате клональной эволюции [4]. Структурные перестройки обычно представлены транслокациями. Идентифицировано более 30 неслучайных транслокаций. Cпецифические хромосомные перестройки являются независимыми диагностическими и прогностическими маркерами, и служат в выборе тактики терапии [1].
В таблице 1 приведён перечень основных молекулярно-генетических аномалий, которые были идентифицированы при ОЛЛ у взрослых и детей и используемых в настоящее время при молекулярной диагностике [1].
15 декабря 2015
- 1172
- 1,0
- 0
- 4
![]()
Юлия Кондратенко

- Генная инженерия
- Генная терапия
- Иммунология
- Онкология
Генетическую инженерию иммунных клеток успешно применили для борьбы с лейкозом. В этой статье рассказывается, какие генетические технологии помогут бороться с тяжелыми болезнями, если общество проникнется к передовым методикам бόльшим доверием.
В начале ноября 2015 года каждое уважающее себя издание написало об успешном излечении от лейкоза годовалой девочки Лейлы Ричардс [1–3]. Острый лимфобластный лейкоз плохо поддается лечению у таких маленьких детей, и обычная химиотерапия помогает им лишь в 25% случаев. Когда стало ясно, что химиотерапия не справилась, родители девочки начали настаивать, чтобы врачи не ограничивались стандартными методиками, а попробовали все возможные способы для ее спасения. Доктора связались с учеными, разработавшими экспериментальную терапию лейкоза, основанную на генетической модификации иммунных клеток. Технология мало того что применяла генную инженерию, к которой с таким опасением относится общественность, так еще и была опробована только на мышах. Членам комиссий по биоэтике и в страшном сне не может присниться одобрение такой методики для массового применения. Но случай Лейлы был исключением, потому что отработанными методами помочь ей уже точно не получалось. Девочке уже нечего было бы терять, и, если бы новый метод не помог ей резко пойти на поправку, мы бы никогда не услышали ее историю, как не слышим истории сотен тысяч других больных раком, которым не помогли никакие методы лечения.
И терапия сработала. Иммунные клетки донора модифицировали таким образом, чтобы они, во-первых, активно атаковали опухолевые клетки того типа рака, что диагностировали у Лейлы, а во-вторых, не причиняли вреда ее здоровым клеткам. Кроме того, модификации генома сделали донорские клетки устойчивыми к лекарствам, которые пациентка для страховки продолжала принимать, хотя они и не действовали на нее достаточно эффективно. Модифицированные иммуноциты донора помогли уничтожить опухолевые клетки и при этом не нанесли вреда здоровым тканям*. Когда все признаки присутствия опухоли пропали, Лейле пересадили подходящий донорский костный мозг, который начал производить новые клетки иммунной системы, сходные с ее собственными. Вновь заработавшая иммунная система уничтожила генетически модифицированные клетки, выполнившие свою задачу.
Этот случай — не первый, когда генное редактирование применяли у людей. Подобные технологии уже использовали в прошлом году, чтобы повысить устойчивость к вирусу иммунодефицита у нескольких ВИЧ-инфицированных [4]. Тот эксперимент тоже оказался удачным и улучшил показатели иммунитета пациентов. Тем не менее первый эпизод не вызвал такого общественного резонанса, как излечение Лейлы. Вероятно, здесь сыграла роль история маленькой больной девочки, которую терапия спасла от смерти буквально в последний момент. В первом же случае не было риска скорой смерти пациентов, которые к тому же были взрослыми.
Можно долго обсуждать, какие компоненты необходимы, чтобы привлечь к истории внимание публики, но в любом случае громкий успех генетической инженерии человеческих клеток очень важен для исследователей, разрабатывающих новые технологии. Благодаря этой счастливой истории, человечество еще на шаг приблизилось к медицине из фантастических романов. Самое интересное, что многие из технологий медицины будущего уже разработаны, но далеко не везде разрешены правительствами, одобрены комиссиями по этике или хотя бы вызывают доверие у простых граждан.
Компания Cellectis, создавшая клетки, которые вылечили острый лимфобластный лейкоз Лейлы, разрабатывает и другие полезные клеточные линии. Каждая из них нацелена на молекулы, характерные для определенного типа рака — к примеру, острого миелоидного лейкоза и миеломной болезни [5]. Сегодня мало кто верит, что возможно создать лекарство от всех видов рака, но Cellectis планирует выпустить целый арсенал иммунных клеток, подходящих для лечения различных его типов. Клеточные линии Cellectis — это наиболее мягкий вариант генно-инженерной терапии человеческих патологий, поскольку такие клетки применяются только для того, чтобы разобраться с проблемой, а затем уничтожаются иммуноцитами пересаженного костного мозга. В этом смысле первый эксперимент по применению генной терапии к ВИЧ-инфицированным выглядит более радикальным: тогда изменяли собственные клетки крови пациентов, которые после модификации и возвращения в кровоток не планировалось оттуда удалять [6]. На самом деле ученые даже рассчитывали, что модифицированные клетки в теле испытуемых размножатся, поскольку будут устойчивы к ВИЧ. Эти ожидания оправдались. Компания Sangamo, разработавшая терапию против ВИЧ, сообщает, что половина из дюжины участников первого эксперимента уже прекратили принимать противовирусные препараты, при этом их уровень Т-клеток, которые раньше атаковал вирус, остается высоким. Сейчас Sangamo дополнительно проверяет свою технологию еще на 70 пациентах [3].
По сравнению с применением модифицированных клеток крови, которые при необходимости можно убрать из организма (что и произошло с модифицированными иммуноцитами после того, как они справились с лейкозом Лейлы), модифицировать клетки в составе органов намного рискованнее. Во-первых, крайне трудно гарантировать, что вирусные векторы, используемые для доставки систем модификации генов, не проникнут куда-то еще, кроме клеток-мишеней. В некоторых случаях модификация лишних клеток может быть опасной: к примеру, если целью являются клетки опухоли, которые нужно заставить прекратить деления, а модифицируются заодно и стволовые клетки, деления которых очень важны для здоровья организма. Другой важный момент — способность вирусных векторов долго оставаться в состоянии боевой готовности. В теории такие модифицирующие агенты могут сохранять активность годами, что не всегда полезно. Наконец, системы генетической модификации, не похожие на родные белки организма, могут вызывать иммунный ответ, что в сочетании с долгим сроком жизни агентов доставки ДНК может привести к хроническому воспалению и другим неприятным патологиям иммунитета.
В общем, биоинженерам еще есть над чем работать, но каждый удачный эксперимент, особенно на людях, повышает доверие общественности к их разработкам, и стимулирует ученых тянуть наш мир в будущее еще активнее.
Читайте также: