
Генетический анализ рака почки

Рак — заболевание, которое ежегодно уносит миллионы жизней, уступая среди причин смертности только сердечно-сосудистым патологиям. Ученые и врачи-онкологи уже давно ведут с ним борьбу, постоянно внедряя новые средства, которые помогают сохранить жизни всё большего числа пациентов. За последние десятилетия поле сражения сильно сместилось с гистологического и клеточного уровня на молекулярно-генетический.
Если раньше было лишь известно, что при раке меняется внешний вид и поведение клеток, то теперь ученые стремятся разобраться в процессах на уровне генов и отдельных молекул. Это стало возможным с развитием молекулярной биологии, и на этом поприще достигнуты немалые успехи.
Рак развивается из-за мутаций, в результате которых эти гены начинают работать неправильно. Генетические дефекты возникают случайно или при воздействии внешних факторов: курения, ультрафиолетового излучения, канцерогенов в пище и окружающей среде. Некоторые мутации (наследственные) человек получает от родителей, другие (приобретенные) — в течение жизни.
Каждый рак уникален, несет собственный набор мутаций. И эти различия могут сильно влиять на прогноз, чувствительность раковых клеток к тем или иным лекарственным препаратам. Выяснить это помогают специальные генетические анализы.
Показания:
Генетические исследования в онкологии помогают решать важные задачи:
Все генетические исследования на мутации, связанные с раком, можно разделить на две большие группы: те, которые проводят у здоровых людей, чтобы выявить риски, и те, которые проводят у онкологических больных, чтобы изучить опухолевые клетки и подобрать правильное лечение. Для каждой группы есть свои показания.
Анализы для людей, у которых уже диагностирован рак
Обычно такие исследования назначают при поздних стадиях онкологических заболеваний, когда стандартные методы лечения не помогают. Эти анализы применяют для диагностики заболевания, подбора персонализированной терапии и оценки прогноза.
Наиболее распространенные исследования из этой группы:
- При меланоме: исследования мутация в гене BRAF.
- При немелкоклеточном раке легкого: гены EGFR, BRAF, ALK.
- При раке толстой и прямой кишки: ген KRAS.
- При раке молочной железы: ген HER2.
- При раке яичников: гены BRCA1, BRCA2.
Эти мутации будут встречаться только в опухолевых клетках. В остальных, здоровых, тканях организма указанные гены будут функционировать нормально.
Анализы для здоровых людей, направленные на оценку рисков
Наследственные мутации человек получает от родителей. Они присутствуют в половых клетках, а значит, их получат все клетки тела человека. В настоящее время с помощью генетического теста можно определить повышенный риск развития следующих типов рака:
- яичников;
- молочной железы;
- щитовидной железы;
- толстой кишки;
- поджелудочной железы;
- простаты;
- желудка;
- почки.
Кроме того, генетические исследования помогают оценить риск меланомы, сарком — злокачественных опухолей из соединительной ткани.
Эксперты из Американского общества клинической онкологии (American Society of Clinical Oncology) рекомендуют рассмотреть возможность проведения генетических исследований на наследственные мутации людям, у которых в семье часто встречались определенные типы злокачественных опухолей, если такой диагноз был установлен у близких родственников. Правильное решение о необходимости обследования помогут принять онколог, клинический генетик.
Что показывает анализ?
Генетические тесты показывают, в каких генах произошли изменения, связанные с повышенным риском рака. Выделяют две группы генов, в которых могут возникать такие мутации.
Распространенные примеры онкогенов — EGFR и HER2. Эти белки-рецепторы встроены в клеточную мембрану. При активации они запускают цепочку биохимических реакций, в результате чего клетка начинает активно, бесконтрольно размножаться. Все мутации в протоонкогенах — приобретенные, они не наследуются.
Европейская клиника сотрудничает с ведущими зарубежными лабораториями. Они применяют современные технологии секвенирования, которые помогают быстро изучить ДНК человека и выявить изменения в сотнях генов:
Существуют ли противопоказания?
Генетические тесты могут нести некоторые негативные эффекты. Когда здоровый человек узнаёт, что у него мутация, связанная с повышенным риском рака, это может стать сильным эмоциональным потрясением. Врач порекомендует рассказать об этом членам семьи, чтобы они тоже знали о рисках, и это может сделать семейную атмосферу более напряженной. Сам по себе генетический анализ стоит недешево. Если его проводят у онкологического больного для подбора персонализированной терапии, рекомендованные по результатам исследования препараты тоже могут оказаться очень дорогими.
Как происходит сдача анализа?
Насколько достоверны результаты?
Точность обнаружения мутаций с помощью современных генетических исследований составляет почти 95%.
Что может повлиять на точность результата?
Для того чтобы анализ показал достоверный результат, врач-онколог должен правильно провести биопсию, соблюдать технику фиксации (специальной обработки) ткани. Организация, которая отправляет материал в лабораторию, должна соблюдать правила транспортировки. В противном случае провести исследование не получится.

Расшифровка анализа
Если анализ на наследственные мутации показал отрицательный результат, это значит, что у человека нет генетических дефектов, повышающих риск развития тех или иных злокачественных опухолей. Но это не значит, что он никогда не заболеет раком. Просто его риски несколько ниже. Аналогично положительный результат не говорит о том, что у пациента обязательно будет диагностировано онкологическое заболевание. У него повышены риски, и, возможно, потребуются некоторые профилактические мероприятия.
Иногда результат исследования на наследственные мутации сомнителен. В таких случаях многие онкологи и клинические генетики предпочитают считать, что риск рака всё же повышен, и рекомендуют некоторые меры профилактики. В ряде случаев ситуацию помогают прояснить анализы близких родственников.
Иногда обнаруживают неизвестные изменения в генах. Непонятно, то ли это вариант нормы, то ли нейтральная мутация, то ли она повышает риск рака.
Если анализ проводится у онкологического пациента для подбора эффективного лечения, лаборатория высылает лечащему врачу отчет, в котором указывает:
- обнаруженные мутации;
- список научных публикаций, в которых эти мутации фигурируют;
- препараты, одобренные для лечения рака с такими генетическими дефектами;
- препараты, которые в настоящее время не одобрены для лечения данного типа рака, но успешно применяются для борьбы с другими злокачественными опухолями с аналогичными мутациями.
На основе этой информации онколог принимает решение по поводу дальнейшего лечения.
Генетические исследования на рак в Европейской клинике
В Европейской клинике есть всё для того, чтобы, при необходимости, назначить онкологическому пациенту персонализированную терапию, замедлить прогрессирование болезни и продлить жизнь. Мы применяем все препараты последних поколений, зарегистрированные на территории России, и сотрудничаем с ведущими европейскими, американскими лабораториями, которые проводят генетические исследования в онкологии.
Мы знаем, как помочь, если в другой клинике сказали, что больше ничего нельзя сделать, или лечение, назначенное ранее, перестало помогать. Свяжитесь с нами.
Злокачественная опухоль почки формируется в эпителии проксимальных канальцев и собирательных трубочек (почечно-клеточная форма) или в эпителии чашечно-лоханочной системы (переходно-клеточная форма). Среди первичных злокачественных образований у взрослых эта онкопатология занимает 80–85 %.
Виды наследственного рака почки
Выделяют такие наследственные синдромы:
- Аутосомно-доминантный поликистоз почек. Повышает вероятность почечно-клеточной карциномы.
- Наследственная папиллярная карцинома. Это семейный синдром наследственного рака почки, а также высокопенетрантная, аутосомно-доминантная патология.
- Болезнь фон Гиппеля-Линдау. Аутосомно-доминантный синдром, сопровождающийся множественными доброкачественными и злокачественными образованиями, включая светлоклеточную карциному.
- Наследственный рак почек, ассоциированный с зародышевыми мутациями цикла Кребса. Это агрессивные формы почечно-клеточной карциномы, метастазирующие даже при небольшом размере и т. д.
Выявление генетических факторов риска по наследственному раку почки у пациента и его семьи позволяет разработать такую стратегию, которая минимизирует или предотвратит болезнь-ассоциированную заболеваемость. Анализу подлежат такие гены/протеины, как c-MET (локус 7q31), фумаратгидратаза (1q42), фолликулин (17p11), сукцинатдегидрогеназа (5p15), TSC1 (9q3416p13), TSC2 (3p25).
Причины рака почки
Основными факторами риска считаются:
- курение;
- ожирение;
- гипертония;
- мужской пол;
- удаление матки;
- тяжелые болезни почек;
- генетические патологии (вызывают наследственные формы рака почки);
- длительный диализ при хронической почечной недостаточности.
Симптомы рака почки
Основными признаками патологии являются:
- гематурия (наличие крови в моче);
- болевой синдром в проекции почки (боль ноющая, тупая, почечная колика);
- задержка мочи (из-за скопления в мочевом пузыре кровяных сгустков);
- повышение артериального давления (вторичная гипертензия);
- пальпируемое образование в области почки;
- симптомы общего характера (потеря веса, субфебрильная температура, нарастающая слабость).
При метастазах в легкие возникает кровохарканье и боль в груди, при поражении печени — признаки желтухи. Метастазирование в кости сопровождается патологическими переломами, в головной мозг — головными болями, радикулитом, неврологической симптоматикой.
Диагностика рака почки
Для обследования используются такие методы:
- УЗИ;
- КТ;
- рентгенография с внутривенным контрастированием;
- МРТ;
- ангиография;
- сцинтиграфия;
- биопсия опухоли с гистологическим исследованием.
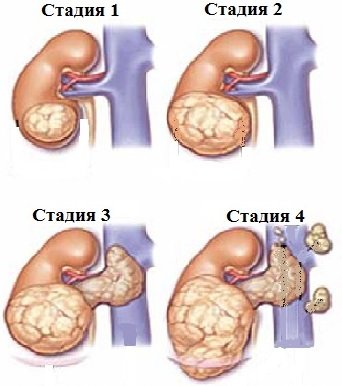
Выделяют четыре стадии рака почки:
- Опухоль ограничена почкой, через капсулу не проникает.
- Образование проникает через капсулу органа.
- Рак почки распространяется на ближайшие лимфоузлы, почечную и нижнюю полую вены.
- Опухоль разрастается на соседние органы (кишечник, поджелудочную железу), появляются метастазы (например, в легких).
Лечение рака почки
Основными методами являются:
Диагностика на ранней стадии онкопатологии позволяет в 90 % случаев добиться излечения.
На правах рукописи
МИХАЙЛЕНКО Дмитрий Сергеевич
АНАЛИЗ МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИХ НАРУШЕНИЙ, АССОЦИИРОВАННЫХ С РАЗВИТИЕМ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ ПОЧКИ.
АВТОРЕФЕРАТ диссертации на соискание ученой степени кандидата медицинских наук
Москва 2008 год
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность темы. Ежегодно в мире регистрируют около 200 тыс. новых случаев рака почки и почти 100 тыс. смертей от этого заболевания, что позволяет считать его одной из основных проблем современной онкоурологии. В России диагностируют до 9 тыс. случаев опухолей почки в год, более 90% которых приходятся на почечно-клеточные карциномы - рак почки (РП). По темпам прироста заболеваемости РП уступает только новообразованиям предстательной и щитовидной желез [Алексеев Б.Я., 2007].
В настоящее время диагностика и прогноз РП основываются на данных инструментальных методов исследования и патоморфологических критериях, что далеко не всегда позволяет правильно оценить прогноз заболевания и возможности лечения [Yin-Goen 2006]. Это обусловливает необходимость создания эффективных тест-систем для проведения своевременной лабораторной диагностики, мониторинга и определения прогноза течения РП. Необходимым этапом создания системы маркеров РП является анализ наиболее характерных и часто встречающихся молекулярно-генетических нарушений в первичных опухолях почки. В качестве таких повреждений генома опухолевых клеток могут выступать мутации, потеря гетерозиготности и аберрантное метилирование ряда генов-супрессоров.
Ген VHL инактивируется в большинстве случаев самого распространенного морфологического типа РП - светлоклеточного рака почки (СРП) вследствие соматических мутаций, метилирования промотора и потери гетерозиготности [Banks R.E., 2006]. Также при СРП выявляют аллельные делеции других генов-супрессоров. Вопрос о влиянии мутаций и метилирования VHL и делеций генов-супрессоров в области 3р (VHL, RASSF1, FHIT) на прогрессию первичной опухоли и прогноз заболевания остается открытым. Идентифицирован ряд генов-супрессоров, аберрантное метилирование которых наблюдается в различных морфологических типах опухолей [Lovisolo J.A., 2006]. Анализ наиболее часто метилируемых генов будет способствовать созданию системы диагностических и прогностических маркеров РП.
Опубликованы данные об исследованиях полиморфизмов различных генов при РП [Karami S., 2008]. Некоторые полиморфизмы могут представлять интерес как потенциальные маркеры предрасположенности к спорадическому РП в популяции европейской части России или влиять на течение заболевания.
РП практически не чувствителен к лучевой и химиотерапии, поэтому хирургическое удаление опухоли является основным методом лечения РП. Определенные успехи связаны с внедрением в практику таргетных препаратов, механизм действия которых напрямую связан с генетическими нарушениями в опухолевых клетках [Costa L.J., 2007]. В связи с этим вопрос о лечении метастатического РП и местнораспространенных форм заболевания требует максимально точной оценки прогноза развития первичной опухоли, знания ее особенностей на молекулярно-генетическом уровне.
Таким образом, комплексное исследование соматических мутаций, потери гетерозиготности, метилирования 5‘-регуляторных областей генов-супрессоров и полиморфных вариантов генов, задействованных в развитии РП, будет способствовать формированию системы новых молекулярно-генетических маркеров РП.
Целью исследования является комплексный молекулярно-генетический анализ при РП, направленный на выявление и характеристику диагностических и прогностических маркеров заболевания.
1. Изучить мутации, аллельные делеции и метилирование промотора гена VHL и провести сравнительный анализ выявленных изменений относительно патологических параметров первичной опухоли и клинических особенностей заболевания.
2. Провести анализ потери гетерозиготности областей локализации генов VHL, RASSF1, FHIT и TP53 в парных образцах РП на различных стадиях заболевания и степенях дифференцировки первичной опухоли.
3. Оценить частоты аберрантного метилирования генов-супрессоров VHL, RASSF1, FHIT, SFRP1 и CDH1 в образцах РП и провести сравнительный анализ метилирования этих генов относительно патологических параметров первичной опухоли и клинических особенностей заболевания.
4. Исследовать метилирование CpG-динуклеотидов в промоторной области гена HOXB13, построить карту аберрантного метилирования этого гена.
5. Определить частоты аллелей и генотипов полиморфных вариантов генов ABCB1, TGFBR1, IL10, VDR в норме и у больных РП. Провести сравнительный анализ полученных данных между группами пациентов и контроля, а также между различными клиническими группами больных РП.
Научная новизна. Идентифицированы 33 новые мутации в гене VHL. Впервые исследованы сочетанные делеции генов VHL, RASSF1, FHIT и ТР53 при СРП. Изучено метилирование 5‘-регуляторных областей генов VHL, RASSF1 , FHIT, SFRP1 и CDH1 с помощью мультилокусной метилчувствительной ПЦР. Впервые показана ассоциация аберрантного метилирования RASSF1 с прогрессией первичной опухоли на ранних стадиях РП (Р = 0.047). С помощью бисульфитного секвенирования построена карта метилирования промотора гена-кандидата HOXB13 в первичных опухолях почки. С помощью нового метода - минисеквенирования с детекцией в режиме фрагментного анализа - изучены полиморфные варианты генов ABCB1, TGFBR1 , IL10 и VDR. Впервые в России определены в норме частоты генотипов исследуемых SNP генов IL10 и TGFBR1, получены данные о возможном влиянии полиморфизмов в генах VDR и TGFBR1 на развитие опухолей почки.
Практическая значимость. Проведено комплексное молекулярно-генетическое исследование 127 первичных опухолей почки (светлоклеточных, папиллярных и хромофобных карцином). Оптимизирован метод комплексной оценки молекулярно-генетических нарушений VHL (соматических мутаций, аберрантного метилирования и потери гетерозиготности) при СРП. Выявлена высокая частота повреждений гена VHL при СРП. Разработаны системы из двух STR-маркеров для тестирования аллельных делеций в областях локализации генов VHL, RASSF1, FHIT и ТР53 при РП. Определена ассоциация множественных аллельных делеций генов-супрессоров на 3р (Р = 0.036), а также метилирования генов RASSF1 и CDH1 с клинико-морфологическими характеристиками опухоли (Р = 0.001), что позволит использовать анализ этих генов в качестве маркеров прогрессии на различных стадиях РП. Результаты, полученные в представленной работе, могут быть использованы в разработке системы молекулярно-генетических маркеров РП, в частности, определение мутаций и метилирования гена VHL - при оптимизации таргетной терапии. Положения, выносимые на защиту:
1. Мутации, потеря гетерозиготности и/или метилирование гена VHL происходят на ранних стадиях СРП в большинстве случаев заболевания.
2. Потеря гетерозиготности двух и более генов-супрессоров, локализованных в области 3р, отражает прогрессию первичной опухоли.
3. Аберрантное метилирование является существенным механизмом инактивации генов-супрессоров VHL, RASSF1, FHIT, SFRP1, CDH1 и наблюдается в 85% первичных почечно-клеточных карцином. Построена карта метилирования промотора гена-кандидата HOXB13 при спорадическом РП.
4. Метилирование генов RASSF1 и CDH1 ассоциировано с прогрессией и метастазированием первичной опухоли, соответственно, что позволяет рассматривать их как составную часть системы молекулярных маркеров РП.
Личное участие автора в получении результатов, изложенных в диссертации. Все эксперименты и методики были разработаны и проведены автором лично. Автор провел статистический анализ всех полученных данных и сформулировал выводы. Описание собственных исследований, анализ и обсуждение результатов выполнены автором самостоятельно.
Публикации. Результаты диссертационной работы отражены в 14 печатных работах соискателя, в том числе, 6 статей опубликовано в журналах,рекомендованных ВАК МОН РФ соискателям ученой степени кандидата медицинских наук.
Объем и структура диссертации. Диссертационная работа изложена на 123 страницах машинописного текста, состоит из оглавления, введения, списка сокращений, обзора литературы, подробного изложения использованных материалов и методов, описания собственных результатов и их обсуждения, заключения, выводов, практических рекомендаций и списка цитируемой литературы, включающего 143 ссылки. Диссертация иллюстрирована 18 таблицами и 18 рисунками.
МАТЕРИАЛЫ И МЕТОДЫ Образцы опухолей почки предоставлены Урологической клиникой им. Р.М. Фронштейна ГОУ ВПО ММА им. И.М. Сеченова, ГУ МРНЦ РАМН и ФГУ МНИОИ им. П.А. Герцена (всего 127 образцов РП). Все случаи РП классифицированы по TNM согласно требованиям Международного противоракового союза (UICC, версия 1997 г.). Из них 53% (67/127) соответствовали I стадии заболевания, 12% (15/127) - II, 21% (27/127) - III и 14% (18/127) - IV. На момент постановки диагноза 17% (21/127) пациентов в изучаемой выборке имели метастазы в регионарных лимфатических узлах и/или отдаленные метастазы.
Геномную ДНК из замороженных образцов опухолей и соответствующих им участков гистологически не измененной ткани выделяли с помощью протеиназы К с последующей фенол-хлороформной экстракцией. Архивные образцы опухолей, заключенные в парафиновые блоки, предварительно депарафинизировали с помощью ксилола и этанола.
Мутации VHL выявляли с помощью ПЦР экзонов 1-3, SSCP-анализа ПЦР-продуктов и последующего секвенирования. При анализе ДНК, полученной из парафиновых блоков, амплифицировали 3‘-часть 1-го экзона.
Для анализа потери гетерозиготности (ПГ) генов VHL, RASSF1, FHIT и ТР53 была разработана оригинальная система из STR-маркеров (по два микросателлитных локуса на каждый ген): D3S1317 и D3S1038 (VHL), D3S1568 и D3S966 (RASSF1), D3S1234 и D3S1300 (FHIT), D17S1353 и IVS1 (TP53). Проводили ПЦР вариабельных локусов, затем ПЦР-продукты разделяли в 10% денатурирующем ПААГ (IVS1-TP53 - в 8% ПААГ).
При подготовке к бисульфитному секвенированию геномную ДНК обрабатывали бисульфитом натрия, который вызывает переход неметилированных остатков цитозина в урацил, но не изменяет метилированные цитозины. Дизайн метилспецифических праймеров был выполнен с помощью интерактивной программы MethPrimer.
Мультилокусную ПЦР полиморфизмов ABCB1, TGFBR1, IL10, VDR проводили с использованием праймеров, фланкирующих исследуемые SNP в каждом из генов (методика разработана в ходе настоящей работы). Не вошедшие в реакцию праймеры и dNTP инактивировали экзонуклеазой I из E.coli и щелочной фосфатазой ("Fermentas", Литва). Далее к ПЦР-продуктам добавляли внутренние праймеры для каждого SNP и проводили реакцию с использованием ABI Prism® SNaPshot TM Multiplex Kit ("Applied Biosystems"), затем проводили обработку продуктов реакции щелочной фосфатазой. Детекцию проводили на генетическом анализаторе ABI PRISM 3100 ("Applied Biosystems").
Статистический анализ частот аллелей и генотипов полиморфизмов, метилирования и ПГ проводили с помощью точного двустороннего критерия Фишера. Комплексный анализ встречаемости генетических нарушений в нескольких группах осуществляли при использовании критерия % .
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ Изучение инактивации гена VHL при спорадическом раке почки
Инактивация гена VHL вследствие молекулярно-генетических нарушений выявляется в большинстве светлоклеточных карцином почки, при этом влияние инактивирующих событий на развитие опухоли неоднозначно. В настоящей работе определяли мутации (рис. 1), аллельные делеции и метилирование как причины инактивации VHL. Мутации VHL были определены в 31.7% (39/123) СРП. Все выявленные мутации соматические, что, наряду с клиническими данными, позволило исключить синдром Хиппеля-Линдау и рассматривать имеющиеся образцы как выборку спорадического СРП. Среди мутаций 69.2% (27/39) составляли делеции, протяженность которых варьировала от 1 до 42 п.н., 18.0% (6/39) - инсерции, дупликации и комплексная мутация, оставшиеся 12.8% (5/39) приходятся на однонуклеотидные замены. Впервые идентифицированные мутации определены в 84.6% (33/39) образцов СРП.
Большинство делеций и инсерций, а также дупликации и комплексная мутация, составляющие 56.4% (22/39), приводили к сдвигу рамки считывания и формированию новых стоп-кодонов.
Рисунок 1. Определение мутации c.472C-G. Вверху - результат SSCP-анализа экзона 3 гена VHL (N и Т - нормальная и опухолевая ткани, соответственно), ПЦР-продукт с аномальной подвижностью в образце 5Т; секвенирование ПЦР-продукта 5Т с обратного праймера, идентификация мутации.
Аллельные делеции VHL при СРП определяли по STR-маркерам D3S1317 и D3S1038. Проанализированы 94 образца замороженных тканей опухолей и соответствующих им образцов нормальной почечной паренхимы, взятых на расстоянии не менее 1.5 см от края новообразования. Информативность используемых микросателлитных локусов составила 47.9% (45/94) для D3S1317 и 77.7% (73/94) - для D3S1038. Система из двух STR-маркеров позволяла определять аллельные делеции в 91.5% (86/94) случаев. ПГ обнаружена в 27.9% (24/86) информативных образцов СРП.
Метилирование промотора VHL исследовали с помощью МЧ-ПЦР (рис. 2). Исследовали 94 парных образца тканей опухолей и почечной паренхимы, а также 29 парафиновых блоков с архивными образцами СРП (всего 123 опухоли). Метилирование промотора гена определено только в образцах СРП и не выявлено в гистологически нормальной ткани. Частота аберрантного метилирования составила 14.6% (18/123) выборки СРП.
Рисунок 2. Анализ метилирования VHL. М - маркер молекулярной массы (pUC19/MspI, размеры фрагментов в п.н. указаны слева), ОК - отрицательный контроль амплификации, ПК - положительный контроль амплификации, К+ - внутренний контроль ПЦР (экзон 2 гена VHL), МЕТ - анализируемый участок промотора VHL, 1-12 - анализируемые образцы опухолей. Аберрантное метилирование выявлено в образцах 5 и 10.
Сопоставлены участки CpG-островка, анализируемого в представленной работе и у других авторов. Показано, что все праймеры для метилспецифической ПЦР (МС-ПЦР) в других исследованиях локализованы внутри участка, изученного в настоящей работе. Один из праймеров всегда содержал CpG-динуклеотиды в позициях -162, -164 и -168, два из которых (-162 и -168) входят в сайты узнавания метилчувствительной рестриктазы BstHHI. Методом МС-ПЦР изучали метилирование 8 остатков цитозина в области с.-24_-168. В настоящей работе проведен анализ метилирования с помощью МЧ-ПЦР семи CpG-динуклеотидов на том же участке. Таким образом, МЧ- и МС-ПЦР могут служить альтернативными методами определения метилирования промотора VHL.
У больных на I стадии СРП соматические мутации в гене VHL выявлены в 35.4% (23/65) случаев, ПГ - в 28.2% (11/39) информативных случаев и аберрантное метилирование - в 20.0% (13/65) образцов. В целом, хотя бы одно из нарушений гена VHL обнаружено у 53.8% (35/65) больных с I стадией заболевания, что свидетельствует в пользу инактивации VHL на ранних стадиях СРП.
Следует отметить, что инактивация VHL влияет не только на развитие первичной опухоли, но и может определять тактику лечения СРП. В последние 3 года в лечении метастатического СРП произошли существенные изменения, и в настоящее время это заболевание можно считать одним из наиболее удачных примеров применения таргетных препаратов. Эти препараты представляют собой ингибиторы определенных тирозинкиназ или факторов роста, которые взаимодействуют лишь с определенными молекулами, играющими ключевую роль в развитии РП. Наиболее эффективные из них - Сутент и Нексавар, ключевые мишени которых (VEGFR 1-го и 2-го типов, PDGFR) гиперэкспрессируются в ответ на инактивацию VHL. Следовательно, СРП, несущий молекулярно-генетические нарушения VHL, может представлять собой наиболее оптимальный случай для терапии одним из этих препаратов. Выдвинутое предположение нашло подтверждение в первом исследовании, показавшем более выраженный эффект применения Сутента у пациентов с соматическими мутациями VHL, чем у пациентов без мутаций [Личиницер М.Р., 2007].
Таким образом, соматические мутации, ПГ и аберрантное метилирование промотора VHL могут рассматриваться как перспективные маркеры СРП. Указанные молекулярно-генетические нарушения присутствуют в первичной опухоли на ранних стадиях заболевания и влияют на чувствительность опухоли к таргетным препаратам.
Исследование аллельных делеций генов VHL, RASSF1, FHIT и TP53
Помимо VHL, для СРП характерны аллельные делеции других генов-супрессоров, которые могут оказывать влияние на развитие злокачественного новообразования. Для определения прогностической значимости аллельных делеций в настоящем исследовании определена ПГ областей локализации генов VHL, RASSF1, FHIT (гены-супрессоры в области 3p) и ТР53 (рис. 3). А 1Т 1Ы 2Т 2N ЗТ ЗЫ Б 4Ы 4Т 5N 5Т БЫ GT
Рисунок 3. Выявление ПГ. А - результат электрофореза в 8% ПААГ пентануклеотидного повтора IVS1 гена ТР53 (T и N - опухолевая и нормальная ткани, соответственно), 1 -гомозигота, 2 - ПГ в опухолевой ткани, 3 - нормальная гетерозигота; В - результат денатурирующего электрофореза в ПААГ микросателлита D3S1568 (образцы нанесены на гель в обратном порядке), 4 - гомозигота, 5 - ПГ в опухолевой ткани (область гена RASSF1), 6 - нормальная гетерозигота.
Информативность STR-локуса D3S1568 составила 58.5% (55/94), D3S966 -62.8% (59/94), D3S1300 - 67.0% (63/94), D3S1234 - 85.1% (80/94), D17S1353 -67.0% (63/94) и IVS1 - 61.7% (58/94), информативность и характеристики систем из двух STR-маркеров для VHL указаны в предыдущем разделе. Системы из двух вариабельных локусов позволяли определять ПГ гена RASSF1 в 85.1% (80/94), FHIT - 95.7% (90/94) и TP53 - 89.4% (84/94) случаев.
ПГ гена RASSF1 обнаружена в 27.5% (22/80), FHIT - 35.6% (32/90), TP53 -17.9% (15/84) и, как упоминалось ранее, VHL - 27.9% (24/86) информативных образцов СРП. Аллельные делеции хотя бы одного гена на 3р наблюдали в 56.4% (53/94), а в совокупности из четырех исследованных генов - в 60.6% (57/94) информативных случаев.

Приглашаем врачей, чья деятельность связана с онкоурологией, к активному сотрудничеству.
Почти каждый десятый случай почечно-клеточного рака имеет наследственный характер. О генетических синдромах, их влиянии на течение болезни и терапию — в системном обзоре на основании последних данных от EAU.
Известны онкологические синдромы, связанные со специфическими мутациями, гистологическим типом опухоли и сопутствующими заболеваниями. Однако ряд наследственных состояний не выдают себя особенностями; заподозрить их можно главным образом по молодому возрасту или семейному анамнезу. Медиана возраста при диагностике наследственного почечно-клеточного рака составляет 37 лет, 70 % опухолей выявляются у пациентов моложе 46 лет. Именно этот возрастной предел повышает вероятность успеха генетического тестирования.
Важное значение имеет анализ наследственных мутаций. Согласно данным Атласа ракового генома (The Cancer Genome Atlas, TCGA), в 6 % случаев светлоклеточного почечно-клеточного рака (сПКР, clear cell renal cell carcinoma — ccRCC) выявляются герминальные мутации, при наличии папиллярного и хромофобного рака их частота равна 9 и 6 % соответственно.
Эта наследуемая по аутосомно-доминантному типу патология характеризуется развитием целого ряда полиморфных опухолей (табл . 1) . Причиной развития синдрома является биаллельная инактивация гена-супрессора опухолевого роста VHL. Поражение почек проявляется наличием кист или светлоклеточного почечно-клеточного рака, обычно мультифокального и билатерального характера VHL-синдром традиционно делится на типы 1 и 2, последний, в свою очередь, — на подтипы (табл 2).
Тактика ведения
Обязательный скрининг пациентов с VHL-отягощенным анамнезом начинается с ретиноскопии в возрасте 5 лет и проведения МРТ головного мозга и брюшной полости каждые 2 года. Необходимы исследование крови и мочи на наличие метанефринов, контроль артериального давления, неврологический осмотр, наблюдение у офтальмолога, отоларинголога и уролога. Генетическая консультация показана пациентам с гемангиобластомами сетчатки глаза и/или ЦНС, лицам молодого возраста и больным с мультифокальными карциномами почек и надпочечников.
Наследуется по аутосомно-доминантному типу. Характеризуется медленным ростом, множественностью, билатеральностью поражения и исключительно папиллярным вариантом 1 типа строения опухоли. В отличие от других синдромов отсутствуют внепочечные проявления. Возраст начала заболевания варьирует; отмечены случаи диагностики болезни у лиц в возрасте 30 лет, к 80 годам фенотипическое проявление приближается к 100 %.
Тактика ведения
Для наблюдения рекомендуются МРТ или КТ с учетом того, что, подобно спорадическому пПКР, наследственные опухоли по большей части гиповаскулярные и при КТ контрастируются лишь на 10-30 единиц Хаунсфилда (HU). На ультразвуковых изображениях поражения часто изоэхогенны по отношению к фоновой паренхиме почки и могут оставаться незамеченными.
Размер опухолевых очагов обычно не превышает 3 см. Выявление активирующих мутаций гена MET привело к испытаниям его ингибиторов. Среди них форетиниб оказался наиболее эффективным у пациентов с герминальными мутациями MET; частичный ответ был получен у половины пациентов с наследственным раком и только в 9 % случаев спорадического пПКР.
Аутосомно-доминантный синдром, характеризующийся поражениями кожи, кистами легких, спонтанным пневмотораксом и многоочаговыми опухолями почек. Причиной синдрома является мутация в гене-супрессоре опухолевого роста FLCN.
У большинства пациентов старше 30 лет развиваются фиброфолликуломы и акрохордоны на лице и верхней части туловища. Двусторонние легочные кисты увеличивают сопутствующий риск пневмоторакса. Примерно у трети пациентов развиваются почечные карциномы различного гистологического строения, большинство из которых имеют промежуточную агрессивность, сравнимую с таковой в случае VHL-синдрома. Опухоли несколько менее агрессивны, чем при других наследственных состояниях, но заболевание не следует рассматривать как доброкачественное, поскольку возможно метастазирование.
Приблизительно 13-34 % пациентов имеют опухоли почек; часто наблюдаются ангиомиолипомы и поликистоз почек По сравнению с болезнью VHL, в рамках которой всегда развивается сПКР, связанные с BHD-синдромом опухолевые образования различаются по гистологическому типу:
- приблизительно 50-67 % опухолей имеют гибридный онкоцитарно- хромофобный тип;
- 23-34 % — хромофобные опухоли;
- 7-9 % — сПКР;
- 3-5 % — онкоцитомы;
- примерно 2 % — папиллярные раки почек.
Тактика ведения
Ежегодное МРТ- или КТ-исследование органов грудной клетки и брюшной полости рекомендовано начинать в возрасте 20 лет. У пациентов без поражения почек МРТ рекомендована каждые 3 года.
Аутосомно-доминантный синдромом, обусловленный мутацией в гене FH. Болезнь характеризуется предрасположенностью к доброкачественному лейомиоматозу кожи и матки, развитием двусторонних папиллярных почечно-клеточных карцином и ранних лейомиосарком матки. В большинстве случаев возникает папиллярный рак 2 типа с градацией ядер по Fuhrman 3-4.
Тактика ведения В качестве скрининга осуществляют:
- ежегодные МРТ-исследования органов брюшной полости;
- наблюдение у гинеколога и дерматолога.
Полиорганный синдром, при котором почти все пациенты имеют ассоциированные кожные проявления, в том числе гипопигментированные пятна, лицевые ангиофибромы и околоногтевые фибромы . У большинства имеются поражения ЦНС (дисплазия коры головного мозга, субэпендимальные узелки и с меньшей частотой — субэпендимальные гигантоклеточные астроцитомы).
Сопутствующие неврологические состояния включают судороги и расстройства аутистического спектра . Другие проявления включают лимфангиолейомиоматоз (ЛАМ) легкого, преимущественно поражающий женщин, миокардиальные рабдомиомы и гамартомы сетчатки.
Рак почки встречается у 1-4 % больных, опухоль часто бывает двусторонней. ПКР классифицируется на 3 типа:
- ТС-связанный папиллярный ПКР;
- Гибридные опухоли (хромофобный рак или онкоцитома);
- Неклассифицированные эпителиальные опухоли.
Тактика ведения
- МРТ органов брюшной полости каждые 1-3 года;
- КТ органов грудной клетки каждые 5-10 лет при отсутствии легочных проявлений;
- МРТ головного мозга;
- Наблюдение у дерматолога, невролога, офтальмолога.
В случаях опухолей высокой жировой плотности или биопсийно доказанных ангиомиолипом с низким содержанием липидов наблюдение является предпочтительным методом до тех пор, пока самая большая опухоль не достигнет в размерах около 4 см, после чего рассматривается селективная ангиоэмболизация . 4-сантиметровый порог основан на исторических данных относительно контроля ангиомиолипом, основанных на более высокой склонности к спонтанному кровотечению.
На основании результатов исследования EXIST-II FDA одобрило эверолимус для лечения ангиомиолипом, связанных с туберозным склерозом.
Это злокачественная эпителиальная опухоль, которая характеризуется развитием параганглиом, феохромоцитом, желудочно-кишечных стромальных опухолей и ПКР. Отличительным гистологическим признаком является обнаружение в цитоплазме атипических клеток вакуолей или хлопьевидных включений, содержащих эозинофильную взвесь.
Распространенность заболевания неизвестна. Небольшая серия случаев показала более ранний возраст начала (средний возраст 30-40 лет, диапазон 15-72 лет) и редкие двусторонние опухоли, которые могут проявлять агрессивное поведение и имеют большой риск развития метастатической болезни.
Таблица 1. Органы-мишени при синдроме Гиппеля—Линдау

Таблица 2. Типы синдрома Гиппеля—Линдау

Тактика ведения
Ведущим методом диагностики является иммуногистохимическое исследование, определяющее потерю экспрессии гена SDH-B. В связи с тем, что что поражение при SDH-дефицитном ПКР является полиорганным, после подтверждения диагноза требуется комплексный междисциплинарный контроль:
- МРТ брюшной полости каждые 2 года;
- анализа мочи и крови на метанефрины;
- анализ на хромогранин А;
- МРТ всего тела (от черепа до таза).
Учитывая возможность метастазирования даже при небольших опухолевых образованиях, ПКР у пациентов с герминальными мутациями гена SDH-B следует лечить аналогично опухолям при синдроме HLRCC. Вероятно, что препараты, подавляющие гликолиз и синтез жирных кислот, также могут использоваться при этих типах рака. В данный момент проводятся клинические испытания вандетаниба в сочетании с метформином.
Наследственный опухолевый синдром, при котором имеется повышенный риск возникновения почечно-клеточного рака. Тип карциномы при этом — светлоклеточный, опухоли могут быть одиночными или множественными, уни- или билатеральными. Это редкая наследственная форма рака почки, достоверно зарегистрированная у 7 семей.
Аутосомно-доминантный синдром, связанный с повышенным риском развития мезотелиомы легких, увеальной меланомы, кожной меланомы, ПКР и, возможно, других злокачественных новообразований.
Взаимосвязь герминальных мутаций гена BAP1 с возникновением злокачественных новообразований была впервые описана в 2011 году, когда исследования выявили повышенную частоту развития меланомы и мезотелиомы. Спустя 2 года было показано, что ПКР является одной из основных опухолей этого синдрома. Однако полный спектр опухолевых поражений, ассоциированных с данным состоянием, является предметом постоянного анализа.
Риск развития ПКР у носителей мутантного гена BAP1 оценивается в 10 %. Из-за небольшого числа зарегистрированных случаев фенотип BAP1-ассоциированного почечно-клеточного рака до сих пор полностью не выяснен. Преимущественно присутствует светлоклеточная карцинома, но есть как минимум 2 опубликованных случая несветлоклеточного рака. Существуют ли носители этой мутации с ранее установленным ПКР, пока неизвестно.
Тактика ведения
Рассматривается аналогично таковой при спорадическом ПКР, хотя зарегистрированы случаи агрессивного течения, поэтому рекомендуются раннее оперативное вмешательство и тщательный скрининг как почечных, так и внепочечных проявлений:
- наблюдение офтальмологом и дерматовенерологом;
- КТ или МРТ органов грудной клетки/брюшной полости.
Является результатом мультигенного наследования (вызывается комбинацией мутаций нескольких генов); каких-либо конкретных генетических аномалии выявлено не было. Гистологически это, как правило, светлоклеточные карциномы, но могут быть и другие варианты. Клиническая картина этих опухолей является типичной и неспецифической.
Тактика ведения
Для пациентов с семейным сПКР рекомендуется проводить генетическое тестирование на VHL, SDH-C, BAP1, TCS1 и TCS2. Если болезнь возникает в раннем возрасте, показано тестирование на SDH- и FH-мутации.
Тактика ведения
Диагностика частично основывается на изучении клинической картины пациента, но наиболее точный результат обеспечивает генетический анализ. Специфического лечения не существует, используется лишь симптоматическая терапия, в том числе и хирургические пособия для удаления новообразований.
Читайте также: