
Генетические синдромы в онкологии
Рак часто наблюдается у членов одной семьи, объективно подтверждена наследственная природа некоторых злокачественных опухолей. Есть точка зрения, что наследственная предрасположенность — самая вероятная причина всех онкологических заболеваний, и только дело времени, чтобы наука точно установила, мутация какого гена за какой конкретный рак отвечает. Но уже сейчас наследственную передачу рака можно прервать.
Если у человека возникло онкологическое заболевание, очень важно выяснить, есть ли в его роду другие случаи злокачественных новообразований. Семьям, в которых имеется более одного такого случая, нужно пройти консультацию врача-генетика, чтобы понять, есть ли в семейной истории основания для подозрений на наследственный характер патологии. Особенно настораживающим признаком будет онкологическое заболевание в нескольких поколениях семьи. Одним из основных методов работы врача-генетика является составление родословных. Другая важная часть медико-генетической консультации — осмотр и опрос пациента: наследственные заболевания нередко проявляются специфическими признаками.
Принципиальным отличием наследственного рака является возможность его прогнозировать путем выявления патогенных мутаций. На первом этапе семьям, в которых имеется более одного случая развития рака, рекомендуется пройти консультацию врача-генетика, по результатам которого можно будет понять, есть ли в семейной истории основания для подозрения на наследственный характер патологии.
Если в процессе консультации возникают подозрения на наследственную природу заболевания, то следующий этап — целенаправленное генетическое тестирование, поиск мутаций, которые могут вызывать конкретное заболевание. Одни исследования позволяют обнаружить изменения в самом гене, другие — в белке, который кодируется измененным геном. Один ген может претерпеть до 300 мутаций.
В последние годы найдены мутации, ответственные за возникновение и развитие рака молочной железы, яичников, толстой кишки и др. Цель генетического тестирования, или скрининга,— выявить риск возникновения заболевания до появления симптомов. Это дает возможность в одних случаях провести своевременное лечение, в других — рекомендовать меры, позволяющие избежать передачи наследственного заболевания потомству. Мутации генов найдены для нескольких видов рака, тесты на некоторые из них уже используют в клинике — например, тесты на рак груди и кишечника.
От предков или не от предков
Все онкологические заболевания имеют генетическую природу, поскольку при раке гены, отвечающие за правильное деление клетки, повреждены. Но в одних случаях имеют место наследственные мутации, а в других — приобретенные. Результатом повреждения (мутации) гена во всех случаях является бесконтрольное неограниченное деление клеток, что и является сутью ракового процесса.
Несмотря на то что онкологические заболевания имеют генетическую природу, только 10–15% из них передаются по наследству. Почему важно знать, наследственный или ненаследственный рак? Потому что если установлена его наследственная природа, то есть выявлена мутация, вызвавшая его, то известен прогноз и понятна тактика в отношении самого больного и его родственников. Особенно отчетливо наследование мутации прослеживается в случаях так называемого семейного рака молочной железы и яичников, при семейном аденоматозном полипозе и различных опухолевых синдромах (Линча — рак толстой кишки, Ли-Фраумени — разнообразные саркомы и др.). Многие люди, сами будучи здоровыми, являются носителями мутаций, приводящих к наследственным заболеваниям. Если носители одной и той же мутации — оба родителя, заболевание становится неизбежным. Генетическое тестирование позволяет это выявить.
Следует подчеркнуть, что наличие мутации не означает заболевания. Мутация может сидеть в гене много лет до того, как начнет развиваться опухоль. Но, зная про мутацию, врачи могут назначить рациональный режим обследования и профилактического лечения.
Например, у женщин—носительниц гена BRCA1 в 95% случаев в течение жизни разовьется рак груди и в 65% — рак яичников, причем часто рак развивается в молодом возрасте, до 50 лет. Это означает, что носительница должна все время находиться под наблюдением, а в некоторых случаях целесообразно ставить вопрос о профилактическом удалении груди и (или) яичников. У всех на слуху история Анджелины Джоли, которая настояла на удалении обеих молочных желез, поскольку у нее обнаружили мутацию гена BRCA1.
Специалисты знают результаты исследования ткани удаленных молочных желез у 54 шведских женщин—носительниц этого гена в возрасте до 51 года. Ни у одной из них обследование не показывало опухоли груди до операции, но гистологическое изучение удаленной ткани выявило наличие раковых клеток у пяти (10%!) из них.
К профилактической хирургии прибегают и при семейном аденоматозном полипозе, при котором вероятность развития рака толстой кишки после 40 лет достигает 100%, и при других онкологических заболеваниях, если установлена онкогенная мутация.
Понятно, что женщины с отрицательным результатом теста на мутации генов BRCA1 и BRCA2 не застрахованы от спорадического рака груди и яичников. Однако вероятность его возникновения несопоставимо ниже, чем у женщин с положительным тестом.
Женщине следует заподозрить у себя предрасположенность к наследственному раку груди, пройти консультацию врача и генетика и генетическое тестирование, если в семье:
— было более одного случая рака груди или яичников по женской линии (у матери, бабушки, тетки, сестер и т. д.);
— заболевание было диагностировано в молодом возрасте (до наступления климакса);
— были случаи рака груди у мужчины;
— были больные c множественными опухолями (например, у одного человека — рак груди, толстой кишки, матки, рак поджелудочной железы и т. д.);
— были случаи двустороннего рака обеих молочных желез или обоих яичников.
Тестирование и его последствия
Генетическое тестирование имеет несколько преимуществ. Отрицательный результат может принести человеку облегчение, избавить от страха ожидания тяжелой болезни, от которой, возможно, погибли его близкие, а также от регулярных обследований, которые должны быть обязательны в семьях с высоким онкологическим риском. Положительный результат дает человеку возможность принимать обдуманные решения о будущем своем и своего потомства.
Сегодня возможна профилактика наследственного рака, то есть возможность не передать от родителей потомству ген, несущий опасную мутацию. Метод, который позволяет это сделать, называется преимплантационная генетическая диагностика (ПГД). Он заключается в следующем: для пары выполняют ЭКО, проводят генетическую диагностику полученных эмбрионов и переносят в матку женщины только те из них, в которых нет онкогенных мутаций. У родившегося ребенка их не будет, а значит, не будет и наследственного рака.
Открытое письмо Анджелины Джоли, New York Times, 14 мая 2013 года
ПГД проводится не на всем эмбрионе, а на нескольких клетках, которые получают путем его биопсии. Доказано, что биопсия не оказывает влияния на здоровье и состояние ребенка. Другими словами, ПГД не снижает частоту наступления беременности и безопасна для будущего ребенка.
Кроме мутаций, отвечающих за развитие рака груди и яичников, установлены мутации, несущие предрасположенность к меланоме, раку желудка, матки, предстательной, поджелудочной и щитовидной железы, толстой и прямой кишки. Если мутация определена и в семье есть люди, которые хотят иметь ребенка, важно, чтобы они знали о возможности предотвратить передачу следующим поколениям этой мутации и связанного с ней рака с помощью ЭКО и ПГД.

Это партнерский материал, подготовленный при поддержке yRisk
Понятие наследственной предрасположенности к различным патологиям широко известно. На сегодняшний день наследственная предрасположенность особенно интересна в контексте формирования опухолей наследственного типа. Зная о наличии такой предрасположенности, можно реализовывать возможности предиктивной медицины и не допустить развития опухоли у пациента в группе риска. Что такое наследственные опухолевые синдромы и как наиболее эффективно диагностировать их в нашей стране, расскажем в этой статье.
По условиям возникновения все виды злокачественных новообразований можно разделить на спорадические и наследственные. Случаи спорадических опухолей составляют подавляющее число во всемирной популяции. По различным данным, от 5 до 10 % составляют случаи наследственных новообразований или наследственные опухолевые синдромы.
Наследственный опухолевый синдром — это состояние, связанное с формированием опухоли в результате наследования мутаций в генах, ассоциированных с онкогенезом. Мутировавшие гены чаще играют контролирующую роль и ответственны за течение клеточного цикла или репарацию ДНК. При этом, в отличие от спорадических опухолей, при наследственных опухолевых синдромах мутации обнаруживаются в генах клеток всего организма с момента формирования зиготы. Опухоль при наследственном синдроме развивается в более раннем возрасте (на 10–15 лет раньше спорадических опухолей), когда наследственный дефект репарации ДНК или течения клеточного цикла будет сочетаться с накопившимися за жизнь соматическими мутациями . Необходимо учитывать, что понятие наследственных новообразований не тождественно понятию семейных опухолей. В первом случае опухоль имеет известную молекулярно-генетическую причину, семейные же опухоли могут ей не обладать, но демонстрировать высокую частоту встречаемости в одной семейной линии.
Широкое распространение наследственных опухолевых синдромов связано прежде всего с постоянным накоплением генетических мутаций в популяции. Большинство опухолей наследственного типа можно рассматривать как патологии с аутосомно-доминантным типом наследования. Носительство мутаций не влияет на фертильность, что позволяет мутациям широко распространяться в популяции.
Еще одной особенностью наследственных опухолевых синдромов является более частое возникновение первично-множественных новообразований.Так, у женщин билатеральные опухоли молочных желез чаще развиваются именно в контексте наследственного рака молочной железы и яичников. По сравнению с опухолями спорадического типа, лечение наследственных опухолевых синдромов носит более длительный характер и требует больших усилий со стороны онкологов.
Одним из наиболее распространенных наследственных опухолевых синдромов является синдром Линча, или наследственный колоректальный рак без полипоза. Частота встречаемости такого синдрома в общей популяции составляет 1:500, а пенетрантность патологии — до 80 %. Согласно Амстердамским критериям II, подозревать у пациента синдром Линча можно по совокупности признаков:
— наличие как минимум трех членов семьи с гистологически подтвержденным колоректальным раком или раком эндометрия, тонкой кишки, мочеточника или лоханки почки, при этом один из членов семьи является родственником первого порядка для двух других;
— исключен семейный аденоматозный полипоз;
— заболевание имеется у представителей как минимум двух последующих поколений;
— как минимум у одного члена семьи заболевание было диагностировано в возрасте до 50 лет.
В нашей стране большое значение имеет высокая распространенность наследственного рака молочной железы и яичников. Среди всех случаев рака молочной железы наследственные составляют 5–10 %, среди случаев рака яичников — 10–15 %. Критерии, определяющие подозрение на наследственный рак молочной железы и яичников, не являются общепринятыми и варьируют в разных странах. Так, критериями NCCN (Национальной онкологической сети США) являются:
— рак молочной железы в возрасте до 50 лет;
— трижды негативный рак молочной железы в возрасте до 60 лет;
— рак яичников или рак поджелудочной железы в любом возрасте;
— рак молочной железы в любом возрасте при наличии в семье случаев рака молочной железы, рака яичников, рака простаты или рака поджелудочной железы;
— рак молочной железы в любом возрасте при наличии еврейских корней в родословной.
Диагностика наследственного опухолевого синдрома у пациентов с уже имеющейся патологией имеет высокую значимость для подбора средств таргетной терапии. Так, например, при выявлении дефектов BRCA1 и BRCA2 возможно назначение препарата Линпарза (олапариб) — ингибитора белка PARP, ответственного за репарацию ДНК.
Наследственным опухолевым синдромом с наиболее широким спектром возможных новообразований является синдром Ли-Фраумени. Этот синдром характеризуется высоким риском формирования злокачественных новообразований в детском и подростковом возрасте. Часто обнаруживаются опухоли молочной железы, головного мозга, надпочечников, лейкозы и другие новообразования. Синдром Ли-Фраумени связан с мутацией в гене TP53. Белок, экспрессируемый им, играет центральную роль в регуляции клеточного цикла. По этой причине синдром Ли-Фраумени ассоциирован с повышением риска возникновения большого количества различных новообразований.
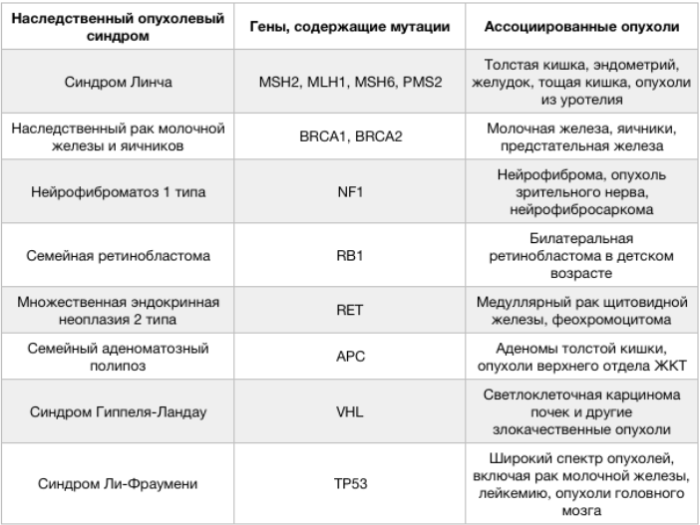
Рисунок 1. Характеристика основных наследственных опухолевых синдромов.
В настоящее время все более широкое распространение в диагностике наследственных опухолевых синдромов получают методы секвенирования. Секвенирование — это процесс определения нуклеотидных последовательностей ДНК и РНК. Методы секвенирования нового поколения (next generation sequencing, NGS) — это целая группа технологий определения нуклеотидных последовательностей ДНК и РНК как в формате отдельных участков генов, так и в объеме целого генома. Определение генетических мутаций методом NGS высокоэффективно: метод позволяет обрабатывать большие объемы данных за короткое время, выявлять и анализировать весь спектр имеющихся мутаций, что снижает число индивидуальных ошибок, связанных с особенностями генома пациента.
В нашей стране генетические исследования методом секвенирования нового поколения проводятся в крупных федеральных центрах, куда можно попасть по направлению врача-онколога. Кроме того, на сегодняшний день любой желающий при подозрении на наследственный опухолевый синдром может пройти генетическое тестирование методом NGS в лаборатории yRisk. Компания работает с использованием современных платформ секвенирования, позволяющих выявлять мутации, ассоциированные с наследственными опухолевыми синдромами. Чувствительность такого анализа значительно превышает данные ПЦР.
Положительный результат исследования не означает наличие у пациента опухоли на данный момент. Обнаружение опухоль-ассоциированных мутаций позволяет включать пациентов в группы риска и регулярно проводить диспансеризацию внутри этих групп. Профилактические мероприятия среди групп пациентов с наследственными опухолевыми синдромами дают возможность предотвратить появление опухоли в раннем возрасте и развитие первично-множественных опухолей, что крайне важно для сохранения высокого качества жизни. Как уже было сказано, генетическое тестирование методом NGS показано и пациентам, уже имеющим опухоль, для подбора наиболее эффективных средств таргетной терапии.
Выявление носительства опухоль-ассоциированных мутаций также позволяет планировать профилактические операции, проведение которых предотвращает развитие опухоли в дальнейшем. Так, при наследственном раке молочной железы и яичников высок превентивный эффект профилактической подкожной мастэктомии. Сохранить качество жизни после этой операции позволяет последующее эндопротезирование молочных желез. Женщинам старше 40 лет с такими мутациями рекомендуется профилактическая овариоэктомия. Профилактические операции рекомендованы и при некоторых других наследственных синдромах.
Диагностика наследственных опухолевых синдромов в современном мире имеет особую значимость. К сожалению, пока не был создан метод, позволяющий эффективно предотвращать развитие спорадических опухолей. Выявление же групп людей с наследственной предрасположенностью позволяет существенно снизить частоту злокачественных новообразований, а в конечном итоге — спасти жизнь не одной тысяче пациентов.
Источники
Parkes A., Arun B. K., Litton J. K. Systemic treatment strategies for patients with hereditary breast cancer syndromes //The oncologist. – 2017. – Т. 22. – №. 6. – С. 655.
Mork M. E. et al. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer //Journal of Clinical Oncology. – 2015. – Т. 33. – №. 31. – С. 3544.
Rahner N., Steinke V. Hereditary cancer syndromes //Deutsches Ärzteblatt International. – 2008. – Т. 105. – №. 41. – С. 706.
В последние годы, с внедрением в клинику молекулярно-генетических исследований, появилось большое количество публикаций об идентификации мутаций в определенных генах, ответственных за наследственную предрасположенность к онкологическим заболеваниям.
Это касается, прежде всего, контингента так называемых раковых семей, в которых среди кровных родственников существует повышенная заболеваемость отдельными формами злокачественных новообразований.
Среди них наиболее хорошо изучены эмбриональные опухоли у детей (ретинобпастома, опухоль Вильмса); семейный аденоматозный полипоз толстой кишки; рак органов женской репродуктивной системы (молочной железы, яичников); синдром Линча и синдром Ли-Фраумени; медуллярный рак щитовидной железы и др. (табл. 10.1).
Таблица 10.1. Некоторые наследственные онкосиндромы, ассоциированные с терминальными мутациями генов-супрессоров или мутаторных генов опухолевого роста (суммированные данные зарубежной литературы). 
Ретинобластоме — классический пример семейного рака — встречается редко, заболеваемость не превышает 3,5 на 1 млн детей. Опухоль развивается в детском возрасте, характеризуется двусторонним поражением глаз, а также повышенным риском развития опухолей молочной железы, легкого и нервной системы в зрелом возрасте.
Возникновение ретинобластомы обусловлено мутацией гена-супрессора Rb1 (хромосома 13q), который играет важную роль в регуляции клеточного цикла.
Ретинобластома наследуется по аутосомно-доминантному типу с высокой пенетрацией. Ее наследственная форма составляет до 40% всех случаев в отличие от большинства других опухолей человека, при которых наследственные варианты составляет не более 1-2%. Носители гена Rb1 часто страдают умственной отсталостью, микроцефалией и другими врожденными уродствами.
Опухоль Вильмса — новообразование детского возраста и семейного характера, часто сочетается с аниридией и врожденными пороками мочеполовой системы. Семейная форма опухоли Вильмса встречается относительно редко (до 30%). Возникновение опухоли Вильмса связано с инактивацией или отсутствием гена-супрессора — WT1. Предполагается, что ген WT1 не единственный маркер семейной формы опухоли Вильмса.
Семейный аденоматозный полипоз толстой кишки
Семейный аденоматозный полипоз толстой кишки (синдром Линча I) во многие десятки раз повышает риск возникновения колоректального рака и который развивается практически у всех больных семейным полипозом.
Синдром сочетается с умственной отсталостью и множественными пороками развития. Причина развития синдрома — инактивация гена-супрессора АРС (adenomatous polyposis coli) на хромосоме 5q.
Носителями терминальной мутации гена являются практически все в семьях с аденоматоэным полипозом толстой кишки. В ряде случаев, как варианты семейного аденоматоза, могут наблюдаться онкогенетические синдромы (Гарднера, Турко, Пейтца-Егерса).
Синдром Линча II семейная форма неполипозного рака толстой кишки
Наследуется по аутосомно-доминантному типу. Синдром Линча II, или синдром множественных аденокарцином, характеризуется семейным распространением первично-множественных аденокарцином ободочной кишки, молочной железы, эндометрия, яичника и опухолей мозга.
Причиной возникновения наследственного неполипозного колоректального рака являются мутация в нескольких супрессорных генах — MSH2 (хромосома 2р), MLH1 (хромосома Зр), PMS2 (хромосома 7р). В норме эти гены играют ключевую роль в процессах репарации ДНК.
Их поломка приводит к накоплению различных генетических повреждений и созданию "мутационного фенотипа", который способствует увеличению вероятности образования злокачественных клеток и клинически проявляется возникновением множественных карцином толстой кишки.
Синдром Ли-Фраумени — сейчас достаточно хорошо изученная онкологическая патология В семьях с этим синдромом повышен риск раннего рака молочной железы и детских опухолей: мягкотканой саркомы, острого лейкоза, опухолей мозга, надпочечников и других органов.
Синдром вызывается зародышевой мутацией в самом изученном супрессорном гене р53 (хромосома 17р). Поэтому обнаружить мутации, ассоциированные с синдромом Ли-Фраумени, относительно несложно.
Семейный рак молочной железы и яичников
Математическими расчетами установлено, что для молочной железы и яичников имеется около 72% общих генов, формирующих предрасположенность к этим двум разным формам рака.
В настоящее время четко установлено, что среди семей, имеющих больных раком молочной железы (в том числе мужчин) и яичников, 5-18% среди них относятся к наследственным вариантам. Развитие наследственного рака молочной железы и яичников связаны с терминальными мутациями генов-супрессоров BRCA1 (хромосома 17q) и BRCA2 (хромосома 13q), идентификация которых названа открытием 90-х годов.
В настоящее время выявлено более 300 вариантов мутаций в BRCA1, которые дают очень высокий риск (44-87%) возникновения рака молочной железы, в большей степени характерного для молодого (до 40 лет) возраста и часто сочетающегося с опухолями яичников.
Мутации в BRCA2 реализуются в раковый фенотип несколько позже; его мутации определяют достаточно значимый риск развития рака яичников (20%); они описаны не только для женщин, но и для мужчин, у которых вызывают рак молочных желез и предстательной железы.
С молекулярно-генетической точки зрения функция генов BRCA1 и BRCA2 (особенно BRCA2) до конца еще не выяснена Однако многие считают, что эти гены представляют собой рецессивные гены-супрессоры, обеспечивающие в норме негативную регуляцию клеточной пролиферации.
Помимо контроля повреждений ДНК и поддержания целости генома BRCA1 связывает рецептор эстрогенов и репрессирует его функцию, регулируя, таким образом, избыточную пролиферацию клеток молочной железы и других эстрогензависимых органов.
Следовательно, при инактивации обоих аллелей BRCA1 пролиферация и злокачественная трансформация эстрогензависимых клеток не сдерживается, что и объясняет, очевидно, возникновение опухолей именно молочной железы и яичника.
МЭН-1 и МЭН-2 синдромы
Различают несколько вариантов наследственного синдрома множественной эндокринной неоплазии (МЭН). Для МЭН 2-го типа (МЭН-2) характерны сочетание медуллярного рака щитовидной железы, феохромоцитомы и других опухолей эндокринных органов. МЭН-2 — единственный наследственный рак, причиной развития которого является мутация не в супрессорном гене, а в классическом (доминантном) протоонкогене RET (хромосома 10q), кодирующем тирозинкиназу.
В отличие от других наследственных раков соматической инактивации второго (интактного) аллеля RET для развития опухоли чаще всего не требуется, так как зародышевая мутация является доминантной на уровне клеток организма, а число возникающих новообразований определяет локализация точковой мутации в RET. При МЭН-1 синдроме также наблюдаются различные опухоли эндокринных желез вследствие мутации гена-супрессора МЭН-1.
К наследственным формам рака относятся болезни Кауденв и Баньяна-Зонана, характеризующиеся множественными гамартомами и обусловленные мутациями супрессорного гена PTEN, а также нейрофиброматоз I и II типов, развивающийся вследствие герминативных мутаций генов NF1 и NF2 (табл. 10.1).
Угляница К.Н., Луд Н.Г., Угляница Н.К.
Об этом мы поговорили с онкогинекологом, хирургом Владимиром Носовым, руководителем Клиники гинекологии и онкогинекологии Eвропейского медицинского центра – первой клиники в России, где персонализированная терапия онкогинекологических заболеваний стала стандартной практикой.
В своём нормальном состоянии эти гены участвуют в восстановлении ДНК после различных повреждений, тем самым защищая клетки от опухолевого перерождения. Если возникает мутация в этих генах, здоровые клетки оказываются не защищенными и сами могут становиться злокачественными. Вероятность заболеть раком груди при носительстве мутации гена BRCA 1/2 колоссальная — до 80%(в общей популяции у женщин без мутации — около 10-12%), риск заболеть раком яичников — до 40-45 %( в популяции —около 1,5%) .
В большинстве случаев назначение этих препаратов после первой линии химиотерапии обеспечивает ремиссию около 3 лет – это огромное достижение, еще никогда в онкогинекологии ремиссия при 3-4 стадии заболевания не продлевалась каким-либо лекарством на столь длительный срок.
Дальнейшие исследования позволили выяснить, что мутации могут быть не только герминогенными, то есть присутствующими во всех клетках организма. Дополнительные 15-20% мутаций генов BRCA происходят только в клетках опухоли, но в крови и других клетках организма их нет. Эти мутации называют соматическими. Они не передаются по наследству, не увеличивают риск развития других онкологических заболеваний, но пациенты, у которых обнаружены мутации в клетках опухоли, также являются кандидатами для лечения ингибиторами PARP.
В Институте онкологии EMC мы предлагаем всем пациентам с раком яичников провести полное секвенирование генов BRCA опухоли и крови. Это позволяет подобрать наиболее эффективную персонализированную терапию. Если речь идет о наследственной мутации – мы рекомендуем в обязательном порядке генетическое обследование детям, сестрам, братьям, родителям, а самим пациенткам-носителям мутации – также пройти дополнительный скрининг на рак молочных желез, риски которого колоссально повышены.
Плохое наследство
Наследственная мутация передается детям с вероятностью 50%, причем как по женской, так и по мужской линии. Носителям мы рекомендуем специальную программу наблюдения и профилактические мероприятия для снижения риска онкологических заболеваний, а также обсуждаем с ними вопросы сохранения репродуктивной функции.
Например, на днях я оперировал пациентку 57 лет с раком яичника. На плановой гистологии был подтвержден злокачественный характер опухоли. Мы провели генетическое исследование опухоли, выявили мутацию BRCA1. Затем было выполнено полное генетическое исследование по крови, чтобы понять, является ли мутация соматической (присутствующей только в опухоли) или герминогенной (наследственной). Выяснилось, что мутация наследственная. Мы рекомендовали пройти обследование двум дочерям пациентки, которые, к сожалению, унаследовали эту мутацию. Женщины-близнецы, им сейчас 31 год, обе еще не планировали беременность и роды. Я рекомендовал им обратиться к репродуктологу, провести стимуляцию и заморозить яйцеклетки, а в 35 лет, именно с этого возраста риски рака яичников начинают расти, удалить профилактически яичники и маточные трубы. В этом случае мы сохраняем матку, и в будущем они смогут выносить своих биологических детей.
Более того, во время ЭКО можно провести предимплантационную диагностику и подсадить эмбрионы, не унаследовавшие мутацию. Таким образом, будущее поколение уже будет защищено.
Рак эндометрия (рак тела матки) – самое распространенное онкогинекологическое заболевание у женщин. Сегодня подходы к его лечению также меняются благодаря персонализированной терапии.
До недавних пор считалось, что существует два типа рака эндометрия. Наиболее частый, первого типа, обычно возникает у полных пациентов, часто с сопутствующими диабетом и гипертонией. Второй – серозный, более агрессивный, не связанный с избытком эстрогенов. На основании клинической картины врачи принимали решение о необходимости дополнительного лечения после операции. Сегодня, благодаря лучшему пониманию биологии опухоли, мы знаем, что этих типов не два, а четыре. И для каждого из них предусмотрено определенное лечение. Чтобы определить, с каким типом рака эндометрия мы имеем дело, достаточно для начала провести иммуногистохимическое исследование.
Каждую опухоль эндометрия вне зависимости от стадии, мы тестируем на наличие определенных молекул, указывающих на благоприятный или менее благоприятный прогноз заболевания. Например, наличие мутации гена P53 говорит о менее благоприятном прогнозе. В этом случае мы рекомендуем не только наблюдение, но и дополнительное лечение с помощью химио-или лучевой терапии.
Некоторые раки матки, так же, как и некоторые раки яичников и молочной железы, имеют в своей основе генетический синдром – синдром Линча. Если мы находим проявления синдрома Линча в опухоли, мы направляем пациентов на полноценное генетическое тестирование. Это важно, потому что рак матки – не единственное заболевание, к которому предрасположены носители мутаций, вызывающих синдром Линча. В частности, у них повышен риск рака толстой кишки в молодом возрасте.
Часто первым возникает рак матки, через какое-то время развивается рак толстой кишки.
Поэтому носителям синдрома Линча рекомендуют начинать скрининг на рак кишки не в 45-50, а гораздо раньше — с 30 лет и делать колоноскопию раз в 6 или 12 месяцев, чтобы не пропустить развитие заболевания.
Выявление синдром Линча у пациентки с раком матки может повлиять и на лечение.
При поздних стадиях пациентам с синдромом Линча мы назначаем специфическую иммунотерапию препаратом пемпролизумаб, что позволяет улучшить прогнозы пациентов.
Генетическое профилирование опухоли – это колоссальный прорыв, который позволил нам подойти к полностью персонализированной терапии в онкологии, основанной не только на диагнозе, но и на понимании биологии опухоли. Для пациентов — это возможность получить точное узкоспециализированное лечение, дающее лучшие результаты, а в случае наследственных раков — возможность защитить будущие поколения от опасных заболеваний.
Читайте также: