
Филадельфийская хромосома при хроническом миелолейкозе
Филадельфийская хромосома характерна для ХМЛ или хронического миелоидного лейкоза, который является клональным новообразованием, развивающимся из кроветворных стволовых клеток.
Данная опухоль была первой, при которой обнаружили признаки характерного хромосомного маркера. Открытие в 1960 году в Филадельфии сделали американские исследователи D. A. Hungerford и P. C. Nowell, в связи в чем этот маркер назвали термином филадельфийская хромосома-Ph. И именно эта находка послужила началом клинической цитогенетики в онкологии.
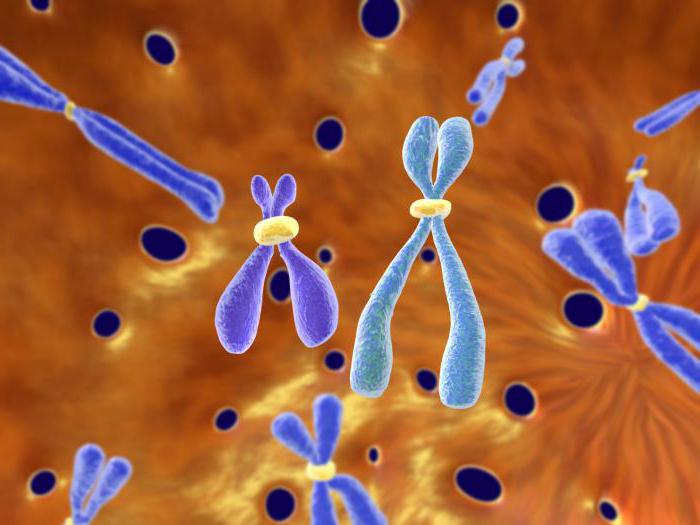
Что собой представляет хромосома?
Может появляться и при других видах лейкоза. Филадельфийская хромосома является укороченной хромосомой, входящей в группу малых акроцентриков. Каждая нормальная женская клетка этой группы содержит их 2 пары– 21 и 22, клетка же мужская включает не четыре, а пять таких хромосом, потому что помимо 21 и 22 пар включается еще и Y-хромосома.
Без G-бендинга, то есть при обычном окрасе, Ph-хромосома выявляется почти у каждого пациента, страдающего ХМЛ, а именно в 95-98 процентах случаев. На хромосомах, которые окрашены дифференциально, видно, что одна из 22 пар является укороченной.
Каков процент обнаружения?
Приблизительно у 90 % пациентов ее видно в каждой анализируемой метафазе, а у остальных больных обнаруживаются и клетки с хромосомой-Ph, и клетки нормальные.
К тому же в некоторых случаях Ph-хромосому регистрируют в меньшинстве клеток костного мозга. Транслокацию (9;22) наблюдают в мегакариоцитах, миелоидных клетках, В- и Т-лимфоцитах, эритробластах. Данный факт является свидетельством того, что болезнь берет начало с предшественницы гемопоэза – некоммитарованной клетки.
Атипичные транслокации
Приблизительно в 10 % случаев можно наблюдать атипичные транслокации, когда цитологическое стандартное исследование может позволить увидеть перенос части 22 хромосомы не на 9, а на любую другую. Более того, в некоторых случаях при ХМЛ обнаруживаются сложные Ph-транслокации при участии не 2 (22 и 9 хромосом), а 3 хромосом или большего количества.

Установили, что почти при каждой Ph-транслокации участвуют 9 и 22 хромосомы, но не всегда это можно увидеть при проведении стандартного цитогенетического исследования, однако это обнаруживают во время использования ПЦР и FISH.
Многочисленные исследователи полагают, что тип Ph-транслокации (сложная, атипичная, стандартная) клинического значения не имеет.
Разрыв генов
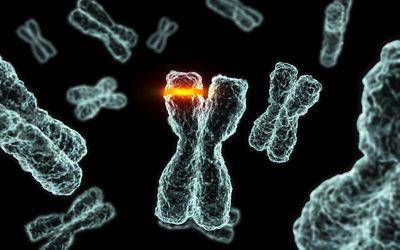
Использование методов молекулярно-генетических поспособствовало установлению того, что разрыв в 9-й хромосоме проходит через протоонкоген (ген ABL), который раньше был идентифицирован у мышей в одном из вирусов лейкоза. В 22-й хромосоме наблюдают разрыв BCR гена. Результатом слияния фрагментов генов BCR и ABL становится образование химерного гена BCR-ABL расположенного обычно на 22-й делетированной хромосоме.
Как вызывает филадельфийская хромосома лейкоз?
Приблизительно 70 % случаев характеризуется обнаружением помимо транскрипта BCR-ABL продукта другого химерного гена, который образуется вследствие t (9;22) —ABL-BCR на der (9), но роль его для развития хронического миелолейкоза не выяснена.
Делеция
Также установили очень важный факт: приблизительно 20-25 % пациентов, у которых обнаружен хронический миелолейкоз, имеют делецию в маркерной 9q+ хромосоме. Эту аномалию нельзя обнаружить при проведении простого хромосомного анализа, но можно увидеть ее во время использования FISH с зондами, которые специально разработаны.
Размер делеции варьируется у каждого пациента: участок с делецией может содержать последовательности BCR-гена, которые перенесены с 22-й хромосомы на 9-ю, на последовательности самой 9 хромосомы или обеих.
Установили также возникновение делеции с формированием специфической t (9;22) одновременно, что частота ее в группах пациентов, которых обследовали на различных стадиях хронического миелолейкоза, одинаковая.
Пока что применение данного метода диагностики (информативного прогностического), к сожалению, в широкую клиническую практику не вошло в связи с его сложностью и требованием дорогостоящего оборудования и реактивов.
Ее роль в прогрессии ХМЛ
Делеция маркера 9q+ играет важную роль в прогрессии ХМЛ, но до конца все еще не выяснена. Когда выпадают кодирующие последовательности генов ABL или/и BCR вследствие делеции, это приводит к экспрессированию лишь одного химерного гена BCR-ABL, но экспрессии гена ABL-BCR не происходит. Вероятно, данное событие также играет важную роль в прогрессировании лейкоза. Более того, исследователи обсуждают возможность инактивации пока еще неизвестных генов-супрессоров, которые локализованы в хромосомном делетируемом районе.
Белок с мол. м. 210 тысяч кодируется химерным геном BCR-ABL, который обладает большей, нежели продукт протоонкогена нормального ABL (H145), протеинкиназной активностью. Во время лейкоза, который у мышей вызван вирусом Абельсона, онкогенную активность имеет белок, продукт gag/abl гибридного гена, который имеет высокую протеинкиназную активность. В эксперименте проводилось вырезание gag/abl гена, после чего вирус уже не мог у мышей вызывать лейкоз. То есть филадельфийская хромосома является маркером этого заболевания.
Подробнее о генах BCR и ABL
При изучении в генах BCR и ABL разрывов при ХМЛ было выявлено, что у разных пациентов разная их локализация. К примеру, в ABL гене протяженность участка, где могут быть разрывы, большая, до 200 kb, а в BCR-гене локализация разрывов происходит на небольшом участке в 8,5kb, то есть можно говорить о наличии кластера разрывов, давшего название самому BCR гену - Breakpoint cluster region.
Во многих случаях t (9;22) разрывы BCR-гена обнаруживаются на участке M-BCR, причем в химерный ген включается длинная часть BCR гена, результатом чего становится появление белка, характерного для ХМЛ P210Bcr/Abl. К тому же при t (9;22) разрывы BCR-гена локализоваться могут на участках, которые называются m-bcr и u-bcr. Bm-bcr области разрывы ведут к образованию P190Bcr/Abl химерного белка, то есть меньшего по величине, чем Р210Bcr/Abl. А локализация разрывов в u-bcr ведет к образованию белка Р230Bcr/Abl более крупного.
Эти различия молекулярного характера коррелируют не строго с клинической особенностью лейкоза. Например, Р190Bcr/Abl имеется у 2 разных видов лейкозов: Ph-позитивный лимфобластный острый лейкоз и гранулоцитарный хронический лейкоз при выраженном моноцитозе и миелопластическими чертами. Если обнаруживают Р230Bcr/Abl, то можно наблюдать картину нейтрофильного хронического миелолейкоза, то есть миелолейкоз, формула крови при котором представлена единичными метамиелоцитами и зрелыми нейтрофилами. К тому же название нейтрофильного гранулоцитарного лейкоза долгое время используют, чтобы обозначать Ph(BCR-ABL)-негативный хронический миелолейкоз (относительно доброкачественный вариант, наблюдаемый у пожилых людей и иногда у подростков).
Любой из вышеприведенных белков можно обнаружить при хроническом миелолейкозе. Также сообщается о достаточно частом сочетании 2 типов белков (Р1900Bcr/Abl и Р210Bcr/Abl) при лимфобластном остром лейкозе и хроническом типичном миелолейкозе.
Роль BCR-ABL для развития хронического миелолейкоза
Решающую роль BCR-ABL гена и его продукта, а именно Р210 белка для развития хронического миелолейкоза демонстрировали на разных системах in vitro и in vivo. Например, при трансдукции bcr/abl в гемопоэтические стволовые клетки мышей и их дальнейшей трансплантации сингенным облученным животным у вторых возникает миелопролиферативная болезнь, похожая на хронический миелолейкоз человека. Установили, что онкогенный потенциал обуславливает высокая тирозинкиназная активность химерного белка BCR-ABL. Одним из центральных в злокачественной трансформации клеток является событие дерегуляции тирозинкиназной активности.
Когда исследователи вводили летально облученным мышам клетки, которые экспрессировали р210Bcr/Abl, то одни животные заболевали лейкозом, сходным с человеческим хроническим миелолейкозом, а другие различными новообразованиями из клеток гемопоэтических: эритроидные опухоли, миеломоноцитарные лейкозы, ретикулоклеточные саркомы, пре В- и Т-клеточные лимфомы, макрофагальные опухоли. Причину различий не выяснили. Такие опыты показывают, что всю цепь событий, приводящую к развитию хронического миелолейкоза еще не установили. Но факт остается. При этом заболевании обнаруживается филадельфийская хромосома в клетках костного мозга. Лечение рассмотрим ниже.
Что еще обнаружили у больных?
Получили данные, которые свидетельствуют о существенном увеличении массы кроветворных клеток и элементов крови в организме пациентов с хроническим миелолейкозом, в основном из-за резкого увеличения срока жизни таких клеток, потому что активированным геном ABL (в BCR-ABL гене) ингибируется апоптоз – запрограммированная клеточная смерть. К тому же этим геном усиливается пролиферация миелоидных клеток.
Имеются основания полагать, что в случае хронического миелолейкоза изменяется функция спец. клеточных белков, то есть интергинов, последствием чего становится нарушение адгезии к стромальным элементам молодых миелоидных клеток, а также происходит избегание стволовых лейкемических клеток негативных регуляторных влияний. Можно сделать вывод. Наличие филадельфийской хромосомы патогномонично для хронического миелолейкоза.
Выводы

Наиболее общей формулировкой молекулярного патогенеза является следующая: химерным геном BCR-ABL кодируется белок, у которого постоянно активирована тирозинкиназная активность, что приводит к активации большого количества сигнальных путей, а также выраженным изменениям апоптоза, адгезии и клеточного цикла. Считается, что эти события достаточны для определения злокачественной клеточной трансформации и поддерживания опухолевого фенотипа.
Но все равно основным маркером является филадельфийская хромосома.
Лечение
Для подавления активности патологических лейкоцитов необходимой для сохранения жизни больных им требуется вторая линия терапии. Для этого применяют нилотиниб – вещество, которое блокирует передачу сигнала от филадельфийской хромосомы.
Ведь именно это и заставляет костный мозг продуцировать в большом количестве поврежденные лейкоциты.
Специалистами выделяется 3 ступени. Во время первой достигается гематологическая ремиссия, нормализация анализа крови и состояния пациента (размера его селезенки).
Но в клетках все же остается филадельфийская хромосома. Во вторую очередь достигается цитогенетическая ремиссия, когда эта хромосома не определяется в клетках. В-третьих, при проведении молекулярного исследования клеток крови в результате воздействия ингибиторами тироксинкиназы у костного мозга не обнаруживают патологический ген.
Трансплантация костного мозга
Обязательна ли ТКМ при филадельфийской хромосоме? Такую операцию по трансплантации проводят больным с острой формой миелоидного лейкоза. Также реально совместимого донора найти бывает непросто. Это очень серьезная и длительная операция. Но она позволяет восстановить нормальную работу костного мозга.
Хронический миелоидный лейкоз (ХМЛ) — клональное новообразование, развивающееся из стволовых кроветворных клеток. Это первая опухоль человека, при которой был обнаружен характерный хромосомный маркер. Открытие сделано в 1960 г. американскими исследователями Р. С. Nowell и D. A. Hungerford в Филадельфии, поэтому маркер был назван филадельфийской (Ph)-хромосомой. Именно с этой находки началась клиническая цитогенетика в онкологии.
Филадельфийская хромосома — укороченная хромосома из группы так называемых малых акроцентриков. В каждой нормальной женской клетке эта группа включает две пары хромосом — 21-ю и 22-ю, а в мужских клетках таких хромосом не 4, а 5, поскольку в эту группу, кроме 21-й и 22-й хромосомных пар, включена также Y-хромосома.
Ph-хромосома выявляется при обычной окраске (без G-бендинга) практически у всех больных хроническим миелоидным лейкозом (95—98 %). На дифференциально окрашенных (бендинг) хромосомах видно, что укорочена одна из хромосом 22-й пары.
Примерно у 90 % больных Ph-хромосома видна во всех анализируемых метафазах, у остальных пациентов обнаруживают как клетки с Ph-хромосомой, так и нормальные клетки, причем в отдельных случаях Ph-хромосома регистрируется в меньшинстве клеток костного мозга. Транслокация (9;22) наблюдается в миелоидных клетках, эритробластах, В-и Т-лимфоцитах, мегакариоцитах. Этот факт свидетельствует о том, что заболевание начинается с некоммитированной клетки-предшественницы гемопоэза.
Примерно в 10 % случаев наблюдаются атипичные транслокации, при которых стандартное цитогенетическое исследование позволяет увидеть перенос фрагмента хромосомы 22 не на 9-ю, а на какую-либо другую хромосому. Кроме того, иногда при хроническом миелоидном лейкозе обнаруживают сложные Ph-транслокации с участием не двух (9-й и 22-й), а трех или более хромосом.
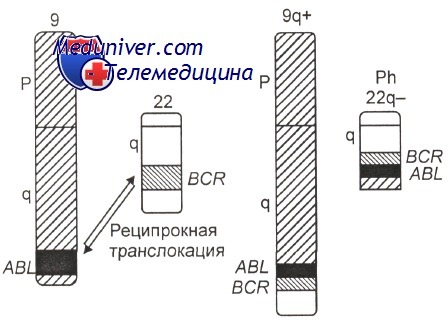
Возникновение химерных генов BCR-ABL и ABL-BCR в результате специфической хромосомной перестройки t(9;22)
Установлено, что практически при всех Ph-транслокациях участвуют хромосомы 9 и 22, однако это не всегда видно при стандартном цитогенетическом исследовании, но обнаруживается при использовании FISH и ПЦР.
Большинство исследователей считают, что тип Ph-транслокации (стандартная, атипичная или сложная) не имеет клинического значения.
Использование молекулярно-генетических методов позволило установить, что в хромосоме 9 разрыв проходит через ген (протоонкоген) ABL, идентифицированный ранее в одном из вирусов лейкоза у мышей. В хромосоме 22 наблюдается разрыв гена ВСЯ. В результате слияния фрагментов названных генов (ABL и BCR) образуется химерный ген BCR-ABL, расположенный, как правило, на делетированной хромосоме 22 (Ph-хромосома) Схематическое изображение молекулярно-генетических событий, приводящих к развитию хронического миелоидного лейкоза, показано на рисунке.
Примерно в 70 % случаев, кроме транскрипта BCR-ABL, обнаруживают продукт (транскрипт) другого химерного гена, образующегося в результате t(9;22) —ABL-BCR на der (9), однако его роль в развитии хронического миелолейкоза неясна.
Установлено, что делеция возникает одновременно с формированием специфической t(9;22), ее частота одинакова в группах больных, обследованных на разных стадиях хронического миелолейкоза. Независимо от метода лечения (даже при использовании программ, включающих гливек) прогноз у больных с интерстициальной делецией в маркере 9q+ существенно хуже, чем у больных без этой аномалии. Длительность хронической фазы и соответственно выживаемость у больных с делецией значительно меньше. Так, при обследовании большой группы больных (241) обнаружено, что медиана выживаемости была 38 мес для больных с делецией (39 человек) и 88 мес для группы больных без делеции (202 человека). Различия статистически значимы.
Применение этого информативного прогностического метода пока, к сожалению, не вошло в широкую клиническую практику, поскольку он весьма сложен и требует дорогостоящих реактивов и оборудования.
Роль делеции маркера 9q+ в прогрессии хронического миелолейкоза пока до конца не выяснена. Выпадение кодирующих последовательностей генов ABL и/или BCR в результате делеции приводит к тому, что экспрессируется только один химерный ген BCR-ABL, но нет экспрессии гена ABL-BCR. Возможно, и это событие играет роль в профессии лейкоза. Обсуждается также возможность инактивации неизвестных пока генов-супрессоров, локализованных в хромосомном районе, который делетируется.
Химерный ген BCR-ABL кодирует белок с мол. м. 210 000, обладающий более высокой протеинкиназной активностью, чем продукт нормального протоонкогена ABL (Р145). При лейкозе, вызванном у мышей вирусом Абельсона, онкогенной активностью обладает белок — продукт гибридного гена gag/abl с высокой протеинкиназной активностью. В эксперименте проводили вырезание гена gag/abl, после этого вирус терял способность вызывать лейкоз у мышей.
Изучение разрывов в генах ABL и BCR при хроническом миелолейкозе показало, что у разных больных их локализация неодинакова. Так, в гене ABL протяженность участка, на котором могут происходить разрывы, велика —до 200 kb, а в гене BCR разрывы локализуются обычно на маленьком участке — 8,5 kb, т. е. имеется кластер разрывов, отсюда название самого гена BCR (breakpoint cluster region).
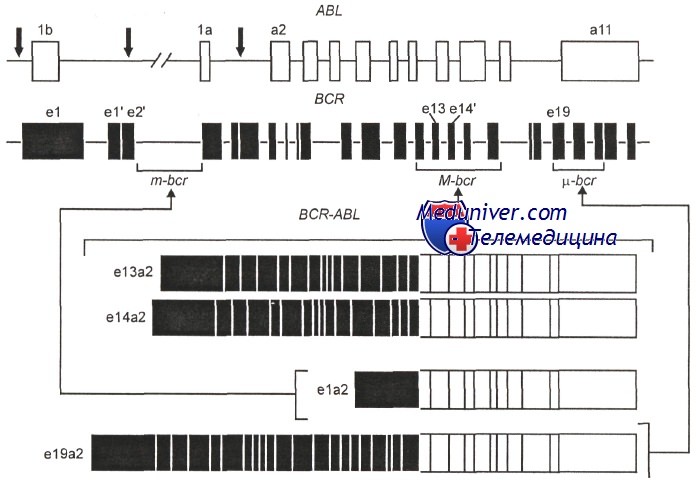
Разрывы в генах ABL и BCR при хроническом миоелоидном лейкозе.
Первый ряд — разрывы в гене ABL; второй ряд — три кластера разрывов в гене BCR: m-BCR, М-BCR и u-BCR. Ниже три типа транскриптов химерных генов BCR-ABL, различающихся по длине вошедших участков гена BCR
В подавляющем большинстве случаев t(9;22) разрывы гена BCR обнаруживают на участке, обозначаемом M-BCR, при этом химерный ген включает длинный фрагмент гена BCR и в результате возникает характерный для хронического миелолейкоза белок P210Bcr/Abl. Кроме того, при t(9;22) разрывы в гене BCR могут локализоваться на одном из двух других типичных участков, называемых m-bcr и u-bcr. Разрывы в области m-bcr приводят к образованию химерного белка Р190Bcr/Abl, т. е. белка меньшей величины, чем Р210Bcr/Abl. При локализации разрывов в u-bcr образуется более крупный белок Р230Bcr/Abl.
Как отмечалось, коррелятивная связь между типом химерного белка (P190Bcr/Abl, Р210Bcr/Abl или Р230Bcr/Abl) и клинико-гематологичесой картиной лейкоза не является строгой. Любой из этих белков может быть обнаружен при классической картине хронического миелолейкоза. Есть также сообщения о нередком сочетании двух типов белков (Р210Bcr/Abl| и Р190Bcr/Abl) при типичном хроническом миелолейкозе и при остром лимфобластном лейкозе.
Решающая роль гена BCR-ABL и его продукта — белка Р210 в развитии хронического миелолейкоза продемонстрирована на разных модельных системах in vivo и in vitro. Так, трансдукция bcr/abl в стволовые гемопоэтические клетки мыши с последующей трансплантацией этих клеток облученным сингенным животным вызывает у них миелопролиферативное заболевание, сходное с хроническим миелолейкозом человека. Установлено, что онкогенный потенциал химерного белка BCR-ABL обусловлен его высокой тирозинкиназной активностью. Дерегуляция тирозинкиназной активности — одно из центральных событий в злокачественной трансформации клеток.
При введении клеток, экспрессирующих р210Bcr/Abl, группе летально облученных мышей в условиях одного и того же эксперимента у одних животных развивался лейкоз, очень похожий на хронический миелолейкоз человека, а у других — самые разнообразные новообразования из гемопоэтических клеток: миеломоноцитарные лейкозы, макрофагальные опухоли, пре-В- и Т-клеточные лимфомы, ретикулоклеточные саркомы и эритроидные опухоли. Причина различий не выяснена. Эти опыты, как и эксперименты с трансгенными мышами, показывают, что вся цепь событий, приводящая к развитию картины хронического миелолейкоза, пока еще не установлена.
Получены данные, свидетельствующие о том, что масса кроветворных элементов и клеток крови в организме больных хроническим миелолейкозом существенно увеличена, главным образом за счет резкого повышения времени жизни этих клеток, поскольку активированный ген ABL (в гене BCR-ABL) ингибирует апоптоз — запрограммированную клеточную смерть. Кроме того, этот ген усиливает пролиферацию миелоидных клеток. Есть основания считать, что при хроническом миелолейкозе изменена функция специальных клеточных белков — интегринов, в результате чего нарушается адгезия молодых миелоидных клеток к стромальным элементам, и стволовые лейкемические клетки избегают негативных регуляторных влияний.

Рубрика: Медицина
Дата публикации: 19.06.2018 2018-06-19
Статья просмотрена: 5100 раз
Филадельфийская хромосома (Ph-chromosome) является наиболее частой цитогенетической аномалией, обнаруживающейся в зрелых В-клетках пациентов с хроническим миелоидным лейкозом (ХМЛ), причем встречаемость этой мутации растет с возрастом, у детей выявляясь в 5 %, а у взрослых — в 50–90 %, достигая максимума у больных в возрасте 35–50 лет [1].
В данном обзоре обобщены результаты работ, опубликованных в системе PubMed и посвященных механизму формирования данной генетической аномалии, а также кратко приведены последствия ее возникновения.
Ключевые слова: филадельфийская хромосома, транслокация, хронический миелолейкоз
Филадельфийская хромосома была открыта в 1960 г учеными из Университета Пенсильвании в Филадельфии (за что и была так названа), в 1973 г в работе Rowley [цит. по 2] было доказано, что она образуется именно путем транслокации. Так, у всех пациентов с ХМЛ в исследовании была обнаружена дополнительная последовательность на длинном плече 9 хромосомы. Это позволило предположить, что Ph-хромосома получается путем переноса части 22ой хромосомы на 9ую (а не простой делеции 22ой хромосомы, как думали изначально) [2].
Транслокация приводит к тому, что участок с онкогеном c-abl, кодирующим тирозинкиназу, переносится к специфическому участку разрыва на 22 хромосоме (breakpoint cluster region — bcr), вместе с тем индуцируя перенос 3'-части гена Абельсона (Abl) с 9ой хромосомы на 22ую вблизи 5'-конца такого же участка bcr [3]. Наиболее важная часть всего процесса — перестановка гена Abl на 22 хромосому рядом с геном bcr и образование химерного гена BCR-ABL [4].
Что касается переноса именно между этими двумя хромосомами, здесь все не так однозначно. Одним из методов диагностики ХМЛ является автоматизированное кариотипирование клеток в стадии метафазы [6], на которой и выявляются атипичные хромосомы (см. Рис.1). Это дает основание полагать, что именно на стадии метафазы, когда хромосомы выстраиваются в экваториальной плоскости клетки, обеспечивается такое взаимное расположение 9ой и 22ой хромосомы, что возникает предпосылка к обмену их участками.
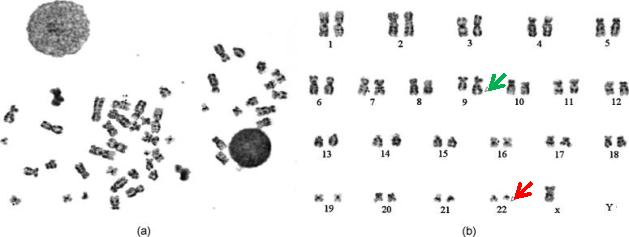
Рис. 1. а) Анализируемая клетка в метафазу и b) ее кариотип. Зеленая стрелка — удлиненная 9ая хромосома, красная стрелка — укороченная 22ая хромосома (Филадельфийская). Иллюстрация из [6] с изменениями.
Нормальный продукт гена BCR — цитозольный фосфопротеин массой 160кДа (р160 BCR ), функция которого не до конца определена. Известно, что первый экзон гена BCR кодирует серин/треонин-киназную активность этого белка, N-концевой домен которого отвечает за формирование димерной структуры, а С-конец обладает ГТФ-азной активностью. Также р160 BCR может быть вовлечен в каскад Ras путем фосфорилирования по тирозину в 177 положении, который таким образом становится способным связывать один из адаптерных белков этого пути [4].
Нормальный продукт гена ABL — тирозинкиназа массой 145 кДа (р145 ABL ), участвующая в процессах клеточного деления, дифференцировки, адгезии и ответа на стрессовые воздействия. С-конец отвечает за связывание с ДНК, а также имеет сайт связывания с актином. На N-конце белка находится три Src-домена: SH3, SH2, SH1. SH3- и SH2-домены регулирующие, а SH1-домен отвечает за наличие функции тирозинкиназы. Мутации в SH2-домене ослабляют связывание фосфотирозина, что приводит к снижению функции белка. SH3-домен имеет депрессорное значение, поэтому именно его удаление превращает белок в онкопротеин, т к его тирозинкиназная активность сильно возрастает [4].
Филадельфийская транслокация дает 3 новых продукта: p190 BCR - ABL , p210 BCR - ABL , p230 BCR - ABL , в каждом из которых нормальные последовательности р145 ABL и р160 BCR соединены голова-к-хвосту [4]. Именно изоформы p210 BCR - ABL обладают повышенной тирозинкиназной активностью и играют огромную роль в развитии хронического миелолейкоза.
Существует 2 изоформы p210 BCR - ABL : b2a2 и b3a2, различающиеся между собой на 25 аминокислот, кодируемых экзоном b3. На настоящий момент нет данных о том, как изоформенный состав связан с различными проявлениями ХМЛ [7].
Конечный продукт химерного гена представляет собой тетрамер, олигомеризация также стимулирует тирозинкиназную активность химерного белка [4].
Помимо этого классического варианта с транслокацией t(9;22), зарегистрированы больные ХМЛ с другим кариотипом. Так, в 1970 г коллективом авторов [цит. по 2] был описан случай 45-летней женщины с типичным ХМЛ. Ей было проведено генетическое исследование, в результате которого было установлено: во всех клетках костного мозга, взятых на исследование, обнаружены филадельфийские 22ые хромосомы, однако 9ые хромосомы во всех образцах интактны (см. Рис.). Это позволило предположить, что не всегда Ph-хромосома образуется одинаково: возможны как варианты с транслокацией t(9;22), так и транслокации между другими хромосомами, а также и простое выпадение фрагментов 22ой хромосомы. Вероятно, таким разнообразием цитогенетических механизмов объясняется гетерогенность течения ХМЛ [2].
Интересно, что несмотря на то, что у пациентов с ХМЛ также может наблюдаться миелофиброз (замещение гемопоэтической ткани костного мозга фиброзной), химерный ген обнаруживается только в клетках гемопоэтической ткани красного костного мозга, и не обнаруживается в фибробластах, что говорит о том, что фиброз является вторичным процессом, не связанным с мутацией напрямую [8].
В результате действия химерного белка нарушаются пути регуляции клеточного цикла, увеличивается пролиферация клеток, их чувствительность к факторам роста, а к проапоптотическим сигналам снижается, так же, как и снижается адгезивная способность к компонентам стромы костного мозга. Все это приводит к тому, что мутантные клетки имеют преимущество в делении (т. к. не чувствительны к апоптозу и быстрее делятся) и вытесняют здоровые клоны, а также выходят в кровь (из-за потери адгезивности) [9]
Почему же эти мутации затрагивают именно гемопоэтические клетки? Возможно, дело в том, что гемопоэтические и стромальные клетки происходят от разных предшественников. В эксперименте [10] было доказано, что стромальные клетки (адипоциты, фибробласты, эндотелиальные клетки и т. п.) и гемопоэтические — это две разные линии. Соответственно, и потенциал генетических изменений у них разный. И, конечно, имеет значение, что данные клетки относятся к быстро делящимся (все ростки включают регулярно обновляемые клетки). Чем быстрее клетки должны обновляться, тем раньше станут заметны их нарушения.
Исходя из этих данных, можно объяснить симптомы ХМЛ. Усталость, истощение, лихорадка являются прямым следствием воспалительных процессов, которые сопровождают любую опухоль; геморрагии отражают тромбоцитопению; боль в костях является следствием неконтролируемой пролиферации клеток в костном мозге; гепато- и спленомегалия, а также лимфаденопатия объясняются заселением этих органов бластными формами; неврологические симптомы возникают вследствие метастазов в головной и спинной мозг. Смерть больных наступает от инфекционных процессов или геморрагического синдрома [11].
7. Wang X., Zheng B., Li S., Mulvihill J. J., Chen X., Liu H. Automated identification of abnormal metaphase chromosome cells for the detection of chronic myeloid leukemia using microscopic images. J Biomed Opt., 2010; 15(4)046026, doi: 10.1117/1.3476336

Что такое хронический миелолейкоз?

Мазок крови пациента с хроническим миелолейкозом
Хронический миелолейкоз (ХМЛ) — злокачественное новообразование кроветворной ткани, сопровождающееся прогрессирующей пролиферацией незрелых гранулоцитов. Заболевание изначально обладает вялотекущим характером, постепенно перетекая в стадию обострения с выраженной симптоматикой и образованием системных нарушений. Является одной из самых опасных и инвалидизирующих болезней.
ХМЛ — первое онкологическое заболевание, у которого определена связь между развитием канцерогенеза и мутацией в гене. Характерная аномалия основана на транслокации 9-й и 22-й хромосом, то есть участки данных хромосом меняются местами, образуя аберрантную хромосому. Выявлена мутировавшая хромосома исследователями из Филадельфии, поэтому она получила название филадельфийская или Ph-хромосома.
Причины развития

Негативное воздействие на кроветворение оказывают ядохимикаты
Заболевание известно науке с 1811 года, но до сих пор факторы, провоцирующие мутацию в гене, определить не удалось. Существует ряд причин, способствующих развитию патологии:
- радиоактивное облучение, в том числе при лучевой терапии;
- химиотерапия иных онкологических заболеваний;
- ряд генетических заболеваний, характеризующихся хромосомной аномалией (например, синдром Дауна);
- взаимодействие с химическими соединениями (нефтепродукты, пестициды).
Патогенез хронического миелолейкоза

Патогенез хронического миелолейкоза
Гибридный ген BCR-ABL 1, образованный в результате транслокации хромосом, продуцирует синтез белка BCR-ABL. Данный белок представляет собой тирозинкиназу, которая в норме способствует передаче сигнальных импульсов для роста клетки. Созданная путём мутации тирозинкиназа становится активным фактором пролиферации клеток, они начинают делиться и распространяться уже независимо от факторов роста. Происходит процесс создания клонов мутировавшей клетки.
Бесконтрольное деление сопровождается нарушением апоптоза — запрограммированной гибели клеток. Также гибридная тирозинкиназа подавляет естественные функции восстановления в молекулах ДНК, создавая предпосылки для последующих мутаций, что усугубляет патологический процесс.
Размножающиеся клетки являются незрелыми, бластными предшественниками полноценных элементов крови. Постепенно бластные клетки вытесняют функциональные эритроциты, тромбоциты и лейкоциты. Добавляются нарушения и в других хромосомах, что запускает ускоренный процесс разрушения организма в целом.
Стадии хронического миелолейкоза

Бластный криз — одна из стадий миелолейкоза
- Хроническая — 30% бластных клеток. Стадия характеризуется агрессивным характером мутировавших клеток, состояние пациента резко ухудшается. Дополнительные аномалии как в гене BCR-ABL, так и в геноме в целом, провоцируют цепь патологических реакций, которые уже практически не поддаются лечению. На этом этапе могут поражаться ткани внутренних органов, кожные покровы и слизистые оболочки, миелоидные клетки преобразовываются в саркому.
Симптомы и признаки

Признаки ХМЛ становятся заметны ближе к прогрессирующей стадии.
- Симптомы опухолевой интоксикации: снижение массы тела, быстрая утомляемость, волнообразное повышение температуры, кожный зуд, тошнота, суставные боли.
- Симптомы опухолевой пролиферации — увеличение селезёнки и печени, боль в левом подреберье, поражение кожных покровов.
- Анемический синдром — головокружение, выраженная бледность, учащённое сердцебиение, чувство нехватки воздуха.
- Геморрагический синдром — склонность к кровоточивости слизистых оболочек, сыпь в виде красных точек, длительное кровотечение при незначительных порезах.
Диагностика заболевания

Один из методов диагностики заболевания — рентгенологический
Диагностика ХМЛ включает:
- Первичный осмотр пациента с изучением анамнеза, жалоб, а также исследование при помощи пальпации размеров селезёнки и печени.
- Общий анализ крови выявляет число и характеристики форменных элементов крови.
- Биохимический анализ проводится для определения уровня билирубина, электролитов, глюкозы, ЛДГ, АСТ, АЛТ.
- Гистологическое исследование костного мозга определяет скопления бластных клеток.
- Цитогенетический анализ выявляет транслокацию хромосом.
- На 3-й стадии проводится иммунофенотипирование для идентификации бластных клеток.
- Метод генного секвенирования применяется для выявления генных мутаций.
- Проводится УЗИ внутренних органов, в первую очередь селезёнки и печени.
- Дополнительно назначают рентгенографию органов грудной клетки, ЭКГ, эхокардиографию, ИФА на маркеры различных заболеваний, коагулограмму и другие исследования.
Лечение

Основа лечения — ингибиторы тирозинкиназы
Выбор препарата и доза определяются в зависимости от стадии ХМЛ и риска побочных эффектов. Обычно лечение начинается с приёма иматиниба в дозировке 400 мг/день при начальной стадии, 600 мг/день при последующих стадиях, затем дозу могут увеличивать или снижать. Различные аберрации в генах обусловливают низкую чувствительность к препаратам, поэтому пациенту могут менять одни ингибиторы на другие.
![]()
Трансплантация костного мозга
Если терапия не оказывает действия, рекомендуется аллогенная трансплантация костного мозга. Новые стволовые клетки могут выработать здоровые элементы кровеносной системы. Но операция сопряжена с рядом высоких рисков.
Терапия препаратами интерферона назначается обычно в 1-й стадии ХМЛ, так как не обладает эффективностью при последующих.
Для уменьшения массы опухоли и при отсутствии результата в лечении ингибиторами проводится химиотерапия. В стадии бластного криза используется полихимиотерапия аналогично лечению острого лейкоза.
Лучевая терапия может быть назначена в случае выраженной спленомегалии. При риске разрыва селезёнки проводят спленэктомию.
Профилактика и прогноз

Прогноз заболевания определяет врач
Причина образования ХМЛ не установлена, поэтому профилактикой являются меры по избеганию контактов с канцерогенными веществами, воздействия радиоактивного облучения.
Прогноз определяется стадией и тяжестью болезни. Одна из прогностических моделей (Kantarjian H.M.) включает факторы:
- преклонный возраст пациента при постановке диагноза;
- концентрация бластных клеток в крови ≥ 3%, в костном мозге ≥ 5%;
- концентрация базофилов ≥ 7%;
- концентрация тромбоцитов ≥ 700*10 9/л;
- выраженная спленомегалия.
Читайте также: