
Экспрессия генов при лейкозах
Традиционно острый миелоидный лейкоз (ОМЛ) рассматривают как результат генетических изменений, ведущих к необратимым дефектам функции критических генов, таким как пролиферация, дифференцировка, апоптоз и транскрипция генов, связанных с лейкозогенезом.
Мутантные гены часто группируют в два класса: гены, которые обеспечивают преимущества роста путем активации прямого воздействия на различные сигнальные пути (включая членов сигнальной трансдукции и активаторов транскрипции (STAT, PI3K и RAS-MAPK), и гены, которые изменяют экспрессию ключевых мишеней транскрипции в миелопоэзе (PML, RUNX1, MLL).
Во всех случаях конечным результатом поражения генов является потеря клетками способности дифференцироваться и отвечать на регуляторы клеточной пролиферации.
Они могут быть преходящими или физиологически необратимыми, играют ключевую роль в развитии эмбриональных структур и в дальнейшем сохраняются на всем протяжении жизни. Эпигенетические изменения, относящиеся к лейкозогенезу, реализуются множественными механизмами, включающими нарушение метилирования ДНК, действие некодирующих РНК и ковалентную модификацию специфических остатков гистоновых протеинов.
Эти протеины являются носителями эпигенетической информации и в комплексе с ДНК образуют структуры протеин/ДНК, называемые хроматином. Для того чтобы ДНК хромосом уместилась в ядре клетки, ее необходимо сконденсировать в компактную структуру (хроматин). Существует несколько степеней конденсации хроматина — от хромосом до нуклеосом. Основной единицей хроматина является нуклеосома, состоящая из основных гистонов: тетрамера Н3/Н4 и двух димеров Н2А/Н2В. Каждый из них представляет собой участок ДНК длиной примерно 200 пар нуклеотидов, который намотан в 2 оборота на белковый октомер.
Посттрансляционная модификация гистоновых протеинов является одним из эпигенетических механизмов в экспрессии ключевых генов при пролиферации и дифференцировке клеток. Эти модификации включают фосфорилирование, ацетилирование, метилирование, окисление дериватов метилированного цитозина и изменения в посттрансляционной модификации гистонов (метилирование лизина), часто в сочетании. Метилирование генов ассоциировано с поддержкой статуса стволовых клеток в гемопоэзе, увеличивая дифференцировку этих клеток; деметилирование наблюдается в генах, участвующих в дифференцировке отдельных клеточных линий.
Гидрометилированная ДНК не способна связывать белки, которые подавляют транскрипцию, результатом чего является ингибирование метилирования ДНК. Лейкозогенез ассоциирован как с гипо- так и с гиперметилированием островков цитозингуанин в различных локусах и с глобальными изменениями метилирования. Ниже представлен механизм эпигенетической дисрегуляции (рис. 2).

Рис. 2. Фузионные протеины при лейкозах и эпигенетическая дисрегуляция.
HDACs — гистондеацетилазы, DNMT — ДНК-метилтрансфераза, HMT — гистон метилтрансфераза, NCOR1 — нуклеарный рецептор ко-репрессора 1, SMRT — закрывающий медиатор ретиноевой кислоты и рецептор тиреоидного гормона, известный также как NCOR2, TF — фактор транскрипции, Ac — ацетилирование гистонов, RNAApolll — РНК полимераза II
Последние исследования в этой области установили, что регулирование генной транскрипции осуществляется двумя путями. В сравнении с нормальными клетками опухолевые клетки обладают гипометилированием ДНК в сочетании с аберрантным метилированием островков цитозин-гуанозин внутри промоторов гена или области кодирования, что ассоциировано с нарушением транскрипционной активности. К тому же обратимые хромосомальные транслокации при ОМЛ приводят к дегенерации фузионных генов, которые в большинстве случаев идентифицируются как регуляторы транскрипции.
Фузионные гены могут также являться регуляторами или медиаторами эпигенетических механизмов. Некоторое количество этих протеинов реализуют лейкозогенез, в частности, путем дисрегуляции транскрипции посредством механизмов, связанных с изменениями хроматина.
Онкогенные фузионные протеины, такие как AML-ETO, CBF-MYH11, PML-RARA, восстанавливают транскрипционный ко-репрессорный комплекс (включающий NCOR1 и SMRT), в результате чего происходит потеря ацетилирования гистонов и появление репрессивной модификации гистонов — метилирование гистона Н3 лизина 9 (Н3К9), триметилирование Н3К27Б, а также метилирование ДНК, что ведет к образованию закрытой структуры хроматина. В итоге это приводит к транскрипционному закрытию различных генов-мишеней, включая гены, которые являются важными для гемопоэтической дифференцировки.
Эпигенетическая или транскрипционная терапия, направленная на фузионные протеины, компоненты корепрессорного комплекса или эффекторы прямой регуляции типа миРНК (микроРНК), является потенциальным резервом изменений, ведущих к ацетилированию гистонов и метилированию Н3К4, открытию структуры хроматина с постепенной транскрипционной активацией и дифференцировкой лейкозного клона.
Понимание механизма аберрантных эпигенетических изменений на молекулярном уровне может предоставить информацию о применении таргетной терапии — ингибиторов малых молекул или специфических ингибиторов энзиматической активности. Эпигенетическая терапия базируется на применении ингибиторов гистондеацетилазы и ингибиторов ДНК-метилтрансферазы (DNMT).
Мутации ДНК-метилтрансферазы ЗА (DNMT3A) типа
Мутации DNMT3A как фермента, участвующего в de novo метилировании цитозин-гуанозиновых динуклеотидов, является наиболее частой соматической мутацией и наблюдается в 15-25% случаев острого миелоидного лейкоза. Мутации DNMT3A выявлены также при МДС и сохраняются после его трансформации в ОМЛ, что согласуется с тем, что они возникают очень рано в процессе клональной эволюции.
Эти мутации найдены более часто при М4/М5 вариантах ОМЛ и ассоциированы с пожилым возрастом, снижением общей выживаемости и конкурентными мутациями FLT3, NPM1 и IDH1/IDH2. Метилирование ДНК обусловлено DNMT, которая катализирует превращение остатков цитозина через гуанозин в 5-метилцитозин путем добавления метильных групп к 5-углеродной позиции цитозина.
Дефицит DNMT приводит к спонтанному дезаминированию тимидина, что ассоциировано с гиперацетилированием гистонов и с появлением открытой конфигурации хроматина, что приводит к активации транскрипции. Метилирование ДНК приводит к стабилизации транскрипционной репрессии и потере функции генов, представленных в промоторах, ассоциированное с активной экспрессией внутри генов в экзонах и интронах.
Эти соотношения метилирования ДНК с экспрессией генов указывают на роль метилирования ДНК в стабилизации нуклеосом и в закрытии сайтов инициации транскрипции. Кроме того, введение метильных групп в ДНК напрямую изменяет связывание с транскрипционными факторами, что ведет к нарушению распознавания внутригенных ДНК-связывающих сайтов в активных экспрессированных генах.
Метилирование ДНК может связывать сайты для протеинов с распознающим метилирование доменом, с индукцией репрессии генов при восстановлении гистондеацетилазы. Аберрантное метилирование ДНК приводит к эпигеномному нарушению структурно нормальных генов, участвующих в регуляции клеточной дифференцировки, пролиферации и жизнедеятельности, что играет роль в патогенезе заболевания.
Гипометилирование может вести к хромосомной нестабильности, мутациям и реактивации разных онкогенов. Все три изоформы DNMT (DNMT1, DNMT3A и DNMT3B) гиперэкспрессируются в бластах в сравнении с нормальными клетками костного мозга. Мутации DNMT3A выявляются у 22,1-27,2% пациентов нормальным кариотипом (НК)-ОМЛ группы промежуточного риска и ассоциированы с неблагоприятным прогнозом в сравнении с пациентами с диким типом DNMT3A, а также с более низкой частотой полных ремиссий. Неблагоприятный эффект мутантной DNMT3A на общую выживаемость более выражен у пациентов с диким типом NPM1 при наличии FLT3-ITD+, FLT3 или мутации CEBPA.
При интегрированном мутационном анализе (J.P. Patel et al., цит. по A.Walker et al., 2012) выявлено, что пациенты, которым проводилась терапия индукции высокими дозами даунорубицина (90 мг/м ), имели более высокую выживаемость при мутантной DNMT3A в сравнении с диким ее типом; поэтому у таких пациентов моложе 60 лет рекомендуется индукцию ремиссии проводить с применением высоких доз даунорубицина.
Авторы рекомендуют у пациентов старше 60 лет при наличии мутации DNMT3A проводить терапию острого миелоидного лейкоза гипометилирующими препаратами (децитабин в виде монохимиотерапии или в сочетании с бортезомибом). Поэтому выявление мутаций DNMT3A у пациентов старшей возрастной группы позволяет выделить группу пациентов для проведения терапии, базирующейся на децитабине.
Мутации изоцитратдегидрогеназы (IDH)
IDH является членом семейства ферментов в-декарбоксилирующих дегидрогеназ, катализирующих оксидативное декарбоксилирование 2,3-изоцитрата до а-кетоглютарата и углекислого газа в цикле Кребса и таким образом участвующим в предупреждении оксидативного повреждения клеток.
Мутации IDH выявлены примерно в 15-30% случаев первичного и вторичного ОМЛ, часто сочетаются с мутациями NPM1 и наблюдаются более часто (до 33% случаев) у пациентов НК-ОМЛ группы промежуточного риска. Они типично гетерозиготные и бывают в трех специфических остатках аргинина — R132 в IDH1 и R172 и R140 в IDH2.
Мутации IDH1 (цитоплазматической) и IDH2 (митохондриальной) при кодировании изоформы протеина с потерей функции приводят к нарушению этих реакций с восстановлением а-кетоглютарата в онкогенную молекулу, 2-гидроксиглютарат, поэтому пациенты с ОМЛ имеют значительно повышенный его уровень.
Имеется несколько механизмов, с помощью которых мутантная IDH может способствовать лейкозной трансформации. Каталитическая активность ТЕТ2 зависит от а-кетоглютарата, железа и кислорода, и поскольку мутация IDH приводит к потере функции ТЕТ2, мутации этих генов взаимосвязаны.
Кроме того, мутации IDH оказывают действие на ТЕТ2-независимые пути лейкозогенеза, влияя на гистондеметилазы посредством других а-кетоглутарат зависимых ферментов. Ингибирование гистондеметилаз способствует метилированию ДНК и может способствовать эпигенетическим нарушениям, выявляемым при лейкозах. К тому же высокий уровень а-кетоглютарата может вызвать оксидативный стресс и привести к повреждению ДНК.
Поскольку практически все мутации IDH можно определить в момент установления диагноза, можно думать, что они появляются очень рано в процессе лейкозогенеза и могут быть кандидатами на инициацию заболевания. Увеличение их количества при вторичном остром миелоидном лейкозе показывает, что они могут быть вовлечены в лейкозную трансформацию и являться пусковым механизмом лейкозогенеза.
Влияние мутантной IDH на прогноз неясно. Хотя не было выявлено разницы в исходе заболевания у пациентов с мутированным и диким типом IDH, общая выживаемость у пациентов моложе 60 лет в группе благоприятного прогноза (NPM1 мутантный/ FLT3 негативный) была короче при наличии мутантной IDH.

Рис. 3. Влияние мутаций IDH при ОМЛ у взрослых
Более высокая общая выживаемость отмечена у пациентов с НК-ОМЛ при наличии мутантной IDH1, IDH2 и мутантным NPM1 в сравнении с пациентами при наличии мутантного NPM1 и диких типов IDH1 и IDH2. Благоприятное влияние мутаций IDH1 и IDH2 на прогноз позволяет выделить дополнительную группу благоприятного прогноза у пациентов с мутацией NPM1.
Мутации ТЕТ2
ТЕТ2 протеин (Ten Eleven Translocation protein2) является деоксигеназой, которая катализирует превращение 5-оксиметилцитозина в 5-гидроксилметилцитозин. Далее он окисляет его до 5-формилцитозина и 5-карбоксилцитозина. 5-гидроксилметилцитозин непосредственно может деметилировать ДНК.
ТЕТ2 часто инактивируется или мутирует при МПН, МДС, хроническом миеломоноцитарном лейкозе и лимфомах, что указывает на его тумор-супрессивную роль. При остром миелоиднм лейкозе мутации ТЕТ2 наблюдаются в 10-23% случаев и являются в большинстве случаев гетерозиготными. Согласно исследованиям, мутации ТЕТ2 более часто наблюдаются при НК-ОМЛ промежуточного цитогенетического риска и ассоциированы с пожилым возрастом, высоким уровнем лейкоцитов, тромбоцитопенией, изолированной трисомией 8, мутациями NPM1 и ASXL1.
Считается, что эти мутации появляются на ранних стадиях лейкозогенеза и могут инициировать опухолевой процесс. Хотя точная роль эпигенетических изменений в результате мутации ТЕТ2 в лейкозогенезе не выяснена, похоже, что обусловленное мутацией ТЕТ2 гидроксиметилирование играет плейотропную роль в модуляции самовосстановления и дифференцировки.
Мутации ТЕТ2 ассоциированы с неблагоприятным прогнозом в группе ОМЛ промежуточного риска, независимо от мутационного статуса гена FLT3. По данным S.Weissmann et al., более короткая бессобытийная выживаемость при мутации ТЕТ2 наблюдается в группе пациентов моложе 65 лет, но не в группе более пожилых пациентов.
Возможно, клиническая вариабельность результатов обусловлена тем, что мутации ТЕТ2 могут происходить в разных регионах гена или же различиями в интенсивности проводимой химиотерапии. Доказано, что потеря функции ТЕТ2 при гематологических новообразованиях приводит в результате к повышению самовосстановления и к нарушению дифференцировки клеток.

Рис. 4. Участие ТЕТ2 в эпигенетической регуляции при остром миелоидном лейкозе у взрослых
Не найдено никакого влияния мутации ТЕТ2 на прогноз при НК-ОМЛ с мутантной NPM1 без мутации FLT3-ITD. Мутации IDH нарушают его ферментную активность вследствие повышения уровня 2-гидроксиглютарата, приводя к глобальному гиперметилированию ДНК, что поддерживает мнение о роли аберрантного гидроксиметилирования в лейкозогенезе.
Мутации гена ASXL1
Ген участвует в регулировании метилирования гистонов, в кооперации с протеином-1 гетерохроматина, модулируя активность гистондеметилазы для Н3К4 и Н3Л9. Соматические бессмысленные, миссенсные, точечные мутации и сдвиг рамки считывания данного гена выявлены в 5-30% случаев ОМЛ.
Мутации в экзоне 12 найдены в 10,8% первичного ОМЛ в целом, в 8,9% при НК-ОМЛ и в 12,9% при остром миелоидном лейкозе с цитогенетическими аномалиями. Модулирующий эффект обусловлен модуляцией эпигенома через активацию и супрессивное взаимодействие с поликомбрепрессивным комплексом и триторакс геном.
Потеря экспрессии гена при ОМЛ ведет к гиперэкспрессии генов-промоторов лейкоза. Кроме того, изменения в геноме из-за неконтролируемой экспрессии НОХ генов ведут к дополнительным мутациям гена ASXL1. Наличие мутаций гена ASXL1 при других МПН говорит о том, что его мутации могут предшествовать мутациям JAK2 и TET2.
Мутации гена ассоциированы с пожилым возрастом, мужским полом, изолированной трисомией 8, мутацией RUNX1 и экспрессией HLA-DR и CD34, но не ассоциируются с t(15;17), комплексным кариотипом, мутациями FLT3-ITD, NPM1 и WT1 и экспрессией CD 15 и CD33. Мутации ASXL1, в частности, сдвиг рамки считывания, ассоциированы с более агрессивным течением, более коротким временем до рецидива и более короткой общей выживаемостью.
В исследованиях J. Chen et al., Pratcorona et al. найдена ассоциация мутации ASXL1 со снижением общей выживаемости пациентов с ОМЛ в группе промежуточного прогноза, хотя при мультивариантном анализе выявлено, что наличие мутаций ASXL1 не является независимым признаком неблагоприятного прогноза.
В целом наличие мутаций в генах DNMT3A, TET2 и ASXL1 может быть ассоциировано с неудовлетворительным прогнозом и эти пациенты иногда представляют отдельный субкласс в группе острого миелоидного лейкоза высокого риска, который требуют более агрессивного лечения. Прогностическое влияние мутаций IDH1/2 пока не ясно.
Группы комплементарных мутаций
При исследовании комплементарного мутационного статуса в когорте Е1900 было выявлено частое сочетание мутаций KIT с изменениями CBF t(8;21) и inv(16)/t(16;16) и достоверное сочетание мутаций IDH1 и IDH2 с мутациями NPM1, а также сочетание мутаций DNMT3A с мутацией аллелей NPM1, FLT3 и IDH1.
Мутации IDH1 и IDH2 наблюдались исключительно без сочетания с мутацией TET2. Детальный мутационный анализ показал, что мутации IDH1 и IDH2 сочетаются исключительно с мутациями WT1. Установлено, что мутации DNMT3A обычно сочетаются с транслокациями MLL.
Научно-исследовательский институт гематологии и трансфузиологии имени Б. Ейвазова, г. Баку, Азербайджан
Острые лейкозы (ОЛ) представляют собой гетерогенную группу опухолевых новообразований системы крови - гемобластозов, характеризующихся первичным поражением костного мозга морфологически незрелыми кроветворными (бластными) клетками с вытеснением ими нормальных элементов гемопоэза. Все острые лейкозы клональны, т.е. возникают из одной мутировавшей кроветворной клетки. [1]. Алгоритм диагностики острых лейкозов в современной клинике основан на пяти базовых компонентах: получение клинических данных, морфологический анализ бластов, цитохимический анализ бластов, иммунофенотипирование бластов, цитогенетическое исследование. В генезе лейкоза лежит нарушение созревания и пролиферации гемопоэтической стволовой клетки, вызывающее блок клеточной дифференцировки и запускающее экспрессию антигенов, которые в норме отсутствуют. Использование метода проточной цитометрии основывается на четком представлении об иммунофенотипических особенностях лейкемических клеток. Определение с помощью линейно-специфичных антигенов иммунологического фенотипа бластных клеток позволяет дифференцировать острые миелобластные лейкозы (ОМЛ), острые лимфобластные лейкозы (ОЛЛ) и смешанно-клеточные лейкозы [2].
Иммунофенотипическая диагностика ОЛ основывается на выявлении фенотипического профиля, практически не встречающегося в норме:
- лейкозов с гетерогенными популяциями бластов;
- лейкозов с аберрантными фенотипами (характеризующиеся отсутствием одного или нескольких линейно-специфичных маркеров);
- лейкозов с асинхронной экспрессией антигенов (характеризующиеся одновременной экспрессией маркеров различных этапов дифференцировки).
Целью нашего исследования явилась оценка экспрессии лимфоидных антигенов на бластных клетках при различных вариантах острых миелоидных лейкозов в республике Азербайджан.
Материалы и методы. Нами было проанализировано 160 больных c диагнозом ОЛ. Среди больных было 55% мужчин и 45% женщин. Медиана возраста составила 55 лет (диапазон 18-75 лет). Разделение на исследуемые группы проводили в соответствии с вариантами лейкоза по ФАБ (франко-американо-британской) классификации. Всего исследовано 7 групп ОМЛ: М0, М1,М2, М3, М4, М5, М6.
Исследование проводилось на 3-х лазерном проточном цитометре FACS CANTO II (Beckton Discinson, USA), с использованием моноклональных антител меченых флюорохромами (FITC, PE, Pecy7, APC, APCcy7, PerCP, Violet) к поверхностным и внутриклеточным дифференцировочным антигенам лимфоидного и миелоидного рядов.
Миелоидные и моноцитарные: CD117, CD13, CD33, CD15, MPO, CD14, CD64, CD11B, CD11C;
Лимфоидный. В-клеточные: CD19, CD22, CD79a, CD10;
Т-клеточные: CD2, CD3, CD5, CD7, CD9, CD4, CD8, CD1a;
Подготовка проб. Материалом служили образцы костного мозга и периферической крови больных, стандартно стабилизированные К2 EDTA. Окраску клеток моноклональными антителами, производили с использованием лизирования с последующей отмывкой клеток.Анализ образцов проводили в программе Facs Diva. Положительной считали экспрессию маркера более чем на 20% клеток. В данном исследовании рассмотрен вариант острого лейкоза с экспрессией маркеров чужих линий, примером которой может быть коэкспрессия лимфоидных антигенов на поверхности бластных клеток у больных острыми миелоидными лейкозами. Выбор аномальных комбинаций антигенов опирался на фундаментальные исследования немецких авторов [3].
Результаты и их обсуждение. Аберрантная экспрессия антигенов дифференцировки чужих линий выявляется в большинстве случаев ОМЛ. При выявлении коэкспрессии маркеров разной линейной принадлежности, является необходимым определение суммарного фенотипа бластов для установления окончательного иммунофенотипического диагноза. Нами было проанализировано 160 случаев острого лейкоза в Азербайджане (рис. 1).
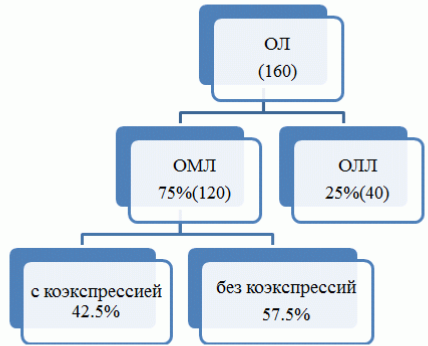
Рис. 1. Структура острых лейкозов в 2014-2015 г. в Азербайджане.
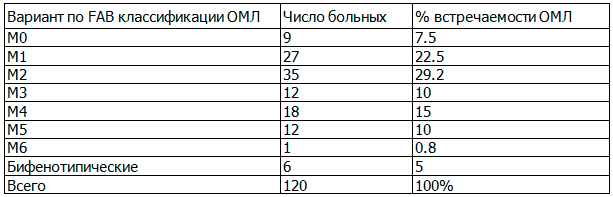
Варианты ОМЛ распределились следующим образом (табл. 1).
Таблица 1. Распределение вариантов ОМЛ в 2014-2015 г. в Азербайджане
Следующим этапом исследования явилось изучение коэкспрессии иммунофенотипических маркеров при остром миелоидном лейкозе. К наиболее частым вариантам аберрантного иммунофенотипа относится коэкспрессия Т-клеточного антигена CD7 у больных ОМ Л. Случаи с экспрессией CD7 на миелобластах характеризуются низкой частотой полных ремиссий и короткой продолжительностью жизни [4].
Объяснением данного клинического феномена может быть связь аберрантного иммунофенотипа с экспрессией гена множественной лекарственной устойчивости MDR1 [5] возможно, что агрессивное течение обусловлено возникновением лейкозного клона из ранних гемопоэтических предшественников. Вместе с тем негативное влияние аберрантной экспрессии CD7 на течение ОМЛ подтверждено не всеми исследователями. Предполагается, что более значимыми для прогноза могут быть другие клинико-лабораторные параметры, нежели иммунофенотипическая характеристика бластных клеток, и прежде всего молекулярно-генетические повреждения [6].
Подтверждением данного предположения может быть связь между кариотипом и иммунофенотипом. Так, у больных с транслокацией t(8;21) на поверхности миелобластов достаточно часто обнаруживается экспрессия антигенов CD34, CD56 и CD7, в случаях с трисомией хромосомы 8 - CD56, а при инверсии inv(16) - CD34 [7]. Аберрантная экспрессия CD7 встречается чаще у больных ОМЛ с промежуточными и неблагоприятными прогностическими вариантами нарушений кариотипа [8]. Анализ протоколов наших исследований выявил СD7 у 30 (25%) пациентов, при этом наиболее часто он встречался при ОМЛ М1-М2 вариантах (рис. 2).
Рис. 2. Проточно-цитометрический анализ образца костного мозга больного ОМЛ М2 с коэкрессией СD7. На I-м дотблоте бокового светорассеяния и CD45 гейтирована бластная популяция. На II-м дотблоте в квадранте Q2 показаны клетки одновременно позитивные по двум антигенам CD7 и CD33.
Антиген CD2 чаще встречается при ОМЛ М3 и М4эоз. Одновременная коэкспрессия антигенов CD2 и CD19 коррелирует с более высокой частотой достижения ремиссий и более длительной 2х летней выживаемостью по сравнению с [CD2;CD19] негативными больными [9]. В нашем исследовании CD2 был выявлен у 12 пациентов (10%) и наиболее часто при ОМЛ М4 варианте. Антиген CD4 встречается на очень ранних стадиях развития клеток-предшественников, коммитированных в грануломоноцитарном и эритроидном направлении, а также на мембране моноцитов и макрофагов. Наличие антигена CD4 чаще обнаруживается у больных ОМЛ с аберрацией [11q23] и коррелирует с низкой выживаемостью [10]. В данном исследовании CD4 был выявлен у 6 пациентов (5%) с диагнозом ОМЛ М5. Ранее экспрессия антигена клеточной поверхности CD9 описана на стволовых кроветворных клетках, эозинофилах, базофилах [11]. Наличие СD9 встречается при всех вариантах ОМЛ, но чаще экспрессия CD9 отмечается при промиелоцитарном лейкозе (рис. 3). В результате наших наблюдений CD9 был выявлен у 12 пациентов (10%), причем почти все случаи ОМЛ М3 были позитивны по CD9.
Рис. 3. Проточно-цитометрический анализ образца костного мозга больного ОМЛ М3v с коэкрессией СD9. На I-м дотблоте бокового светорассеяния и CD45 гейтирована бластная популяция. На II-м дотблоте в квадранте Q2-1 показаны клетки одновременно позитивные по двум антигенам.
Наличие антигена CD10 ассоциируется с более высокой частотой полных ремиссий и большой общей выживаемостью больных. В нашей практике СD10 был выявлен в 6 случаях (5%).
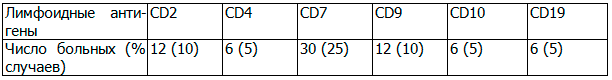
Обобщая данные протоколов наших исследований по экспрессии неродственных антигенов при миелоидных лейкозах, в 42.5% случаев ОМЛ была выявлена лимфоидная позитивность. Частота встречаемости антигенов, ассоциированных с В-линией была значительно меньше, чем антигенов специфичных для Т-линии (таблица 2).
Наиболее часто встречаемый неродственный маркер при ОМЛ М1-М2 вариантах был CD7, при ОМЛ М3 - CD9, при ОМЛ М4 - СD2 и CD4.
Самый высокий процент встречаемости среди лимфоидных маркёров, экспрессируемых одновременно с миелоидными, подтверждён для CD7. Экспрессия CD9 встречалась почти во всех случаях ОМЛ М3. В результате исследования была выявлена высокая частота экспрессии на миелоидных бластных клетках лимфоидных антигенов, из которых преобладающее значение имеют антигены асоциированные с Т-клеточной линией дифференцировки. Аберрантная коэкспрессия В-лимфоидных и миелоидных маркеров встречается в 5.5 раза реже, чем коэкспрессия Т-лимфоидных и миелоидных антигенов.
Таблица 2. Общая частота экспрессии Т-или В-клеточных антигенов у больных ОМЛ
По рекомендациям ВОЗ (2008), в диагностике гемобластозов методом проточной цитофлоуриметрии должны предусматриваться не только возможности диагностики первичного варианта опухоли, но и минимальной остаточной болезни (МОБ) [12].
Выявление у больных аберрантной экспрессии, позволяющей отличать бластные клетки от нормальных гемопоэтических клеток, дает возможность определения резидуальных лейкемическик клеток.
Исходя из вышеизложенного, необходимо отметить значимость выявления такого рода аномальных экспрессий для определения суммарного фенотипа бластов с целью улучшения иммунофенотипической диагностики острых лейкозов, более полной оценки ремиссии и риска развития раннего рецидива заболевания.
В последние годы благодаря использованию новых биологических технологий сделан определенный шаг вперед в понимании патогенеза хронического лимфолейкоза (ХЛЛ), хотя многие моменты остаются неясными. В возникновении заболевания, как и при других лимфоидных опухолях, основную роль, по всей вероятности, играют длительная антигенная стимуляция и генетические нарушения.
Первый вопрос, возникающий при изучении опухоли, — из каких нормальных клеток она возникла.
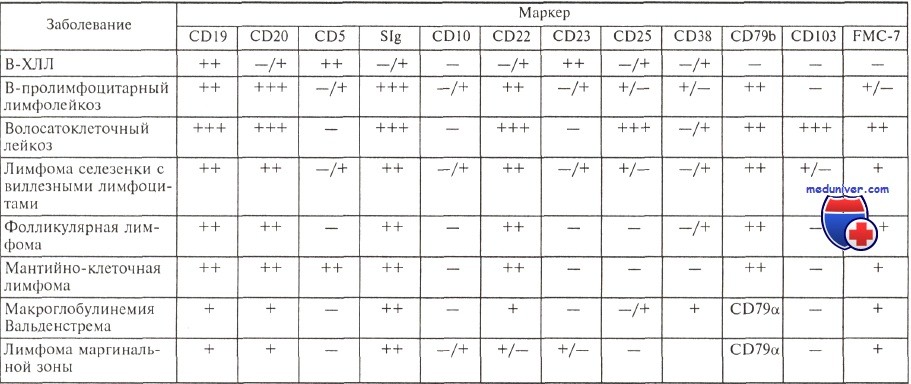
Морфология лимфоцитов при хроническом лимфолейкозе (ХЛЛ) соответствует нормальным зрелым В-лимфоцитам, а их иммунофенотип — иммунофенотипу клеток зоны мантии вторичного фолликула лимфатического узла: сочетание экспрессии CD5 и слабой экспрессии поверхностных моноклональных иммуноглобулинов — IgM, нередко одновременно с IgD и очень редко с экспрессией IgG или IgA.
Нормальные CD5+-лимфоциты обнаруживают еще у новорожденных, они участвуют в независимом от Т-лимфоцитов иммунном ответе — продукции полиреактивных низкоаффинных антител, главным образом антител к собственным антигенам и полисахаридным микробным антигенам. Данные изучения аминокислотной последовательности вариабельного района Н- и L-цепей иммуноглобулинов этих лимфоцитов свидетельствуют о том, что нормальные CD5-позитивные В-лимфоциты не имеют признаков соматических гипермутаций или обнаруживают лишь очень небольшое число мутаций генов, кодирующих образование вариабельных районов иммуноглобулинов (генов IgV). Это также соответствует представлениям об их участии в независимом от Т-лимфоцитов иммунном ответе. У взрослых таких лимфоцитов значительно меньше, чем у новорожденных.
Они составляют примерно 10 % лимфоцитов костного мозга и около 30 % лимфоцитов лимфатических узлов и сосредоточены в зоне мантии вторичных фолликулов. На основании этих данных до последнего времени принималось без сомнений, что хронический лимфолейкоз развивается из клеток мантии вторичного фолликула лимфатического узла. Затем было показано, что клетки маргинальной зоны лимфатических узлов и селезенки человека имеют все черты лимфоцитов при В-ХЛЛ, кроме экспрессии CD5, которая, однако, может появляться после активации этих клеток. Кроме того, клетки маргинальной зоны также участвуют в выработке аутореактивных антител. Есть мнение, что, возможно, именно клетки маргинальной зоны служат источником развития хронического лимфолейкоза.
Сравнение интенсивности экспрессии различных антигенов на поверхности В-лимфоцитов при хроническом лимфолейкозе показало, что лимфоциты с признаками мутаций IgVH-генов интенсивно энспрессируют антигены CD71, CD62L, CD39, на поверхности клеток без признаков таких мутаций высока плотность экспрессированных антигенов CD38, CD69, CD40, HLA-DR.
Корреляция между мутационным статусом генов IgV и экспрессией CD38 обнаружена еще в конце 90-х годов. Оказалось, что CD38 экспрессирован на поверхности лимфоцитов в большинстве случаев ХЛЛ без признаков мутаций IgVHн-генов и отсутствует на большинстве лимфоцитов, имеющих признаки таких мутаций. CD38 является трансмембранным гликопротеином, экспрессированным на многих пролиферирующих гемопоэтических клетках. В недавно вышедших работах показано, что молекула CD38, обладающая энзиматической активностью и катализирующая обмен никотинамидов в лимфоцитах и поступление кальция, при ХЛЛ играет роль рецептора, проводящего сигнал пролиферации и дифференцировки клеток.
Кроме того, CD38 функционирует как молекула адгезии, обеспечивая связь клеток, на которых она экспрессирована, с эндотелиальными клетками. Все эти данные доказывают участие молекулы CD38 в пролиферации патологических лимфоцитов при ХЛЛ. P. Ghia и соавт. показали, что в тех случаях, когда у одного и того же больного имеются популяции CD38-позитивных и CD38-негативных клеток, CD38-позитивные лимфоциты статистически значимо чаще и в большем количестве обнаруживаются в костном мозге, чем в крови. Это согласуется с частым началом прогрессирования или рецидива заболевания с увеличения лимфоцитарной инфильтрации костного мозга.

Известно, что в клонах лимфоцитов с различным мутационным статусом активны разные гены, кодирующие Н-цепи иммуноглобулинов. Всего существует более 200 таких генов (IgVH-гены), однако идентифицированы как функционально активные (кодирующие образование белка) только 44 гена. Эти гены разделены на 6 семейств — от VH1 до VH6.
Все авторы, исследовавшие мутационный статус при ХЛЛ, указывают, что при хроническом лимфолейкозе без признаков соматических мутаций IgVH-генов чаще других функционируют гены 1-го семейства — VH1-69, в случаях с мутациями IgVH-генов — либо гены VH4-34, либо VH3-21. В Японии при обследовании 101 больного установили, что преобладающими по частоте функционирования являются гены семейства VH4 даже в группе без мутаций IgVH-генов. При исследовании 33 больных только у одного из 12 с отсутствием мутаций обнаружили функционирование генов VH1-69, в то время как в сообщениях из других стран функционирование этих генов выявили в 20— 30 % случаев.
В семье с несколькими больными хроническим лимфолейкозом преобладающими оказались гены семейства VH3 как в случаях с мутациями, так и без них. Пока неясно, отражает ли эта находка особенность семейных случаев вообще или присуща только этой семье.
Мутационный статус не меняется на протяжении болезни пациента.
С помощью ДНК-микрочипов было показано, что профиль экспрессируемых генов при хроническом лимфолейкозе независимо от мутационного статуса резко отличается от профиля экспрессируемых генов при других В-клеточных лимфомах и генов, экспрессируемых нормальными В-лимфоцитами. Обнаружено, что при хроническом лимфолейкозе с различным мутационным статусом имеется разница в экспрессии примерно 200 генов из изученных 14 000. Оказалось, что экспрессия тирозинкиназы Zap-70 в 93 % случаев совпадает с мутационным статусом, она достоверно чаще экспрессирована в лимфоцитах больных с отсутствием мутаций IgVH-генов.
Причины различной экспрессии Zap-70 у больных хроническим лимфолейкозом (ХЛЛ) с различным мутационным статусом лимфоцитов неясны. Zap-70 экспрессирована в нормальных Т-лимфоцитах и натуральных киллерах. В Т-лимфоцитах Zap-70 фосфорилируется и проявляет тирозинкиназную активность при передаче Т-клеткам сигнала активации через Т-клеточный рецептор. Роль Zap-70 в В-лимфоцитах пока неясна. Недавно в одной из работ показано, что при хроническом лимфолейкозе имеется корреляция между экспрессией Zap-70 в Т- и В-лимфоцитах: экспрессия Zap-70 в Т-лимфоцитах больных ХЛЛ оказалась значительно более выраженной, чем ее экспрессия в Т-лимфоцитах здоровых доноров (р = 0,001). Добавление фитогемагглютинина к культуре вызвало увеличение экспрессии Zap70 как в Т-, так и в В-лимфоцитах. Это позволяет высказать предположение, что первичная активация Т-лимфоцитов индуцирует экспрессию Zap-70 В-лимфоцитами.
Если эти данные будут подтверждены, роль Т-клеток в патогенезе одного из вариантов ХЛЛ станет более очевидной.
В 2005 г. появились работы, в которых показано, что, помимо различий в экспрессии Zap-70, патологические лимфоциты при хроническом лимфолейкозе (ХЛЛ) различаются по экспрессии других белков. Оказалось, что лимфоциты с различным мутационным статусом достоверно различаются по наличию или отсутствию экспрессии липопротеинлипазы (LPL) и металлопротеиназы ADAM29.
Липопротеинлипаза играет центральную роль в метаболизме и транспорте липидов низкой плотности. Мутации гена, кодирующего ее образование, приводят к дислипидемии и развитию атеросклероза. LPL экспрессирована на клетках жировых тканей, сердечной мышцы, скелетных мышц, тканях молочной железы в периоде лактации. Она не эксперссируется нормальными В- и Т-лимфоцитами. Металлопротеиназа ADAM29 относится к семейству трансмембранных белков, регулирующих взаимодействие клеток друг с другом и с окружающими тканями. Она также не экспрессируется нормальными лимфоцитами. Причины экспрессии этих протеаз частью В-лимфоцитов при хроническом лимфолейкозе неизвестны.
Оказалось, что общая и безрецидивная выживаемость больных различаются в зависимости от наличия или отсутствия экспрессии LPL. Сопоставление экспрессии LPL и мутационного статуса показало, что в 84 % экспрессия LPL обнаруживается у больных с отсутствием мутаций IgVH-генов, а отсутствие экспрессии LPL — у больных с мутациями этих генов. Одновременное определение экспрессии LPL и ADAM29 позволило получить еще более близкое совпадение с мутационным статусом. Отношение LPL/ADAM29 более 1 в 90 % случаев совпадало с отсутствием мутаций IgVH-генов, менее 1 — с их наличием. Это показывает, что мутационный статус обусловливает профиль экспрессируемых на клеточной мембране белков, определение которых может использоваться вместо трудоемкого определения мутационного статуса.
Таким образом, к настоящему времени известно, что между двумя вариантами хронического лимфолейкоза имеются существенные биологические отличия. Тем не менее пока нет данных, позволяющих судить о том, одинаковые или разные по своему происхождению лимфоциты являются субстратом развития хронического лимфолейкоза при этих вариантах. Это могут быть одинаковые клетки, но малигнизация могла произойти на разных уровнях их дифференцировки, однако малигнизироваться могли и лимфоциты разных подгрупп. В последнее время все чаще высказывается предположение, что при обоих вариантах хронического лимфолейкоза малигнизируются клетки маргинальной зоны, экспрессирующие на своей поверхности IgM и небольшое количество IgD. Лабораторные исследования показывают, что эти клетки реагируют на антиген без участия Т-лимфоцитов.
Обычно лимфоциты, пролиферирующие в результате Т-независимой антигенной стимуляции, имеют немутировавшие IgVH-гены, однако имеются доказательства, что клетки маргинальной зоны могут проходить циклы соматических гипермутаций вне терминального центра. Эта гипотеза хорошо согласуется с похожим генным профилем обоих вариантов хронического лимфолейкоза.
Известно, что примерно у трети больных хроническим лимфолейкозом имеются мутации гена BCL-6, одного из регуляторов клеточного цикла. Мутации BCL-6 происходят в терминальном центре. Они обнаружены только в лимфоцитах, имеющих мутации IgVH-генов, что заставляет предполагать единый механизм их возникновения в терминальном центре вторичного фолликула лимфатического узла. В то же время мутации BCL-6 обнаружены только у трети больных с мутациями IgVH-генов, поэтому возможно, что в части лимфоцитов соматические гипермутации происходят вне терминального центра.
Таким образом, пока нет данных, позволяющих с полной уверенностью говорить о том, какая популяция нормальных лимфоцитов является источником развития хронического лимфолейкоза. С несомненностью доказано существование двух вариантов заболевания, однако неизвестно, почему при одном из них после встречи с антигеном в лимфоцитах не произошли гипермутации IgVH-генов и какова в этом роль В-клеточного рецептора.
Читайте также: