
Злокачественная эпилепсия синдром веста

Такая форма эпилепсии, также именуемая инфантильными спазмами, зачастую появляется на 4 — 6 месяцах жизни. Мальчики болеют чаще, чем девочки.
В период приступа происходит быстрый наклон тела вперед, голова сгибается. Судороги появляются, когда грудничок засыпает, или перед пробуждением. Частота приступов может достигать и сотен в сутки.
Причины, которые вызывают синдром Веста
Наиболее распространенная причина синдрома — гипоксическое повреждение мозга ребенка во время тяжелых родов.
Причины появления синдрома Веста таковы:
- врожденные нарушения развития головного мозга;
- различные генетические болезни;
- асфиксия;
- у недоношенных грудничков – кровоизлияния внутри черепа.

На фото ребенок с синдромом Веста
Бывает, что причину этой болезни определить невозможно. В таких случаях следует говорить об идиопатической форме синдрома.
Пациенты с клиническими признаками без изменений в энцефалограмме или больные без признаков, но с изменениями на ЭЭГ, относятся к первой группе риска. Они не нужно лечение, но необходимо ежегодное обследование.
Больные второй группы имеются умеренно выраженные клинические и ЭЭГ-признаки.
Они подвергаются лечению и должны проходить обследование дважды в год. К третьей группе риска причисляют пациентов с яркими клиническими и электроэнцефалографическими проявлениями.
Признаки и симптомы
Синдром Веста имеет такие признаки:
- частые, особо не поддающиеся лечению эпилептические припадки;
- гипсаритмия – стандартные для синдрома изменения на ЭЭГ;
- нарушение психомоторного развития.
У 90% детей с этим нарушением приступы начинаются на первом году жизни, чаще всего в промежутке от 3 до 8 месяцев.
Вначале доктора определяют колики, т.к. признаки и плач, который издает ребенок во время и после припадка, очень похожи на колики.
Стандартными признаками являются:
- наклоны тела вперед;
- судороги тела, рук и ног;
- руки и ноги могут разводиться в стороны в одинаковой степени для правой и левой стороны тела.
Чаще всего каждый приступ продолжается одну или две секунды, затем наступает короткая пауза, после которой наступает следующий спазм. На видео вы можете увидеть, как синдром Веста проявляется у маленького ребенка.
Притом, что у малышей могут наблюдаться единичные спазмы, инфантильные приступы при синдроме, как правило, идут чередой из нескольких спазмов подряд.
Детям с этим нарушением свойственны раздражительность, торможение развития и даже деградация, которые не исчезают без лечения. Такие груднички могут также вести себя так, будто они не видят.
Состояние здоровья нормализуется, когда заболевание начинают лечить и данные ЭЭГ улучшаются.
Лечебные процедуры
Основными способами лечения являются назначение стероидных препаратов или вигабатрина.
Стероиды нужно принимать осторожно, потому что они приводят к неприятным побочным эффектам. Вигабатрин также имеет подобное свойство.
Нейрохирург может осуществить операцию по рассечению спаек мозговых оболочек, удалить опухоль мозга и врожденные аневризмы сосудов. Применяется высокоточная методика вмешательства посредством стереотаксической хирургии, щадящие эндоскопические способы.

Хорошие результаты получены при лечении синдрома Веста стволовыми клетками. Способ еще новый и пока дорогой, но явно перспективный.
Суть лечения состоит в восстановлении нарушенного участка мозга посредством базовых стволовых клеток, являющихся основой для любой ткани, даже для нервной.
Для лечения идиопатической формы нарушения применяются лекарственные препараты:
- противосудорожные средства (нитразепам, эпилим);
- гормональные и стероидные препараты (преднизолон, гидрокортизон, тетракозактид);
- витаминотерапия – большие дозы витамина В6 (пиридоксин).
Эффективность лечения определяется по тому, как быстро исчезают припадки и снижается их выраженность. Если медикамент подобран грамотно и доза его достаточна, то ребенок начинает правильно развиваться и обучаться в будущем, вести полноценную жизнь.
К сожалению, препараты не всегда дают эффект и даже наиболее новые.
Также необходимо учитывать, что у лекарств есть побочное действие:
- чувство постоянной усталости;
- нарушение концентрации;
- кожные и эндокринные реакции;
- депрессия;
- поражение периферических нервов;
- отклонения в работе печени.
Чем больше используемая доза, тем сильнее выражаются побочные эффекты.
В ситуации с симптоматической формой заболевания прогноз и сегодня остается довольно тяжелым.

Подбираем самые эффективные триптаны от мигрени и изучаем специфику действия препаратов на организм.
Стоит ли принимать лекарство Альгерика — инструкция по применению, отзывы и другая полезная информация о лекарственном препарате.
Прогноз, летальность и продолжительность жизни при синдроме Веста
Невозможно предугадать общий прогноз развития из-за разных причин, а также различных симптомов. Каждый случай необходимо анализировать отдельно.
Прогноз для грудничков с идиопатическим синдромом Веста более оптимистичен, нежели для симптоматических форм.
В случае идиопатического синдрома наблюдается менее серьезная задержка развития до начала припадков, а сами судороги легче лечить. У детей с этой формой зачастую происходит преобразование в иные формы эпилепсии и приблизительно 40% детей не отличаются от здоровых.
В иных случаях лечение синдрома имеет трудности, и результаты неудовлетворительные, для малышей с симптоматическими формами прогноз негативный, особенно, если выявлена невосприимчивость к медикаментам.
По исследованиям при синдроме Веста продолжительность жизни 5% детей с нарушением не доживают до 5 лет, в некоторых случаях именно из-за болезни, в некоторых – из-за осложнений лечения.
Менее 50% достигают ремиссии посредством медикаментов. Статистика доказывает, что только 30% случаев лечатся нормально, и только каждый 25-й малыш имеет относительно нормальный уровень физического и умственного развития.
До 90% малышей страдают задержкой физического или умственного развития, даже при удачном лечении судорог. Зачастую это имеет отношение не к самим припадкам, а к причинам, находящимся в основе их развития. Тяжелые постоянные припадки могут сами стать фактором повреждения мозга.
Постоянные повреждения мозга, связанные с синдромом Веста, включают в себя когнитивные нарушения, трудности в обучении, поведенческие отклонения, паралич, отклонения в психике и аутизм.
Около 60% детей с синдромом болеют эпилепсией в дальнейшей жизни. Иногда нарушение преобразовывается в иные формы. Приблизительно половина всех случаев становится синдромом Леннокса – Гасто.
Пятая часть больных детей умирают до года. Причиной смерти являются врожденные патологии развития мозга. Из выживших, три четверти страдают от отклонений психомоторного развития.
Крайне важно вовремя выявить синдром Веста и сразу же начать соответствующее лечение. Чем раньше начато лечение, тем больше вероятность позитивного исхода заболевания.
При правильном подборе противоэпилептических препаратов в половине случаев получается добиться абсолютного излечения ребенка от приступов.
Поздняя диагностика и неправильный выбор лекарств приводят к потере драгоценного времени и ухудшают прогноз болезни.

- Общая информация
- Причины развития
- Клинические проявления
- Диагностические мероприятия
- Подходы к лечению
- Прогноз
Общая информация

Синдром Веста: симптомы и лечение
Заболевание носит вторичный характер, так как в 90% случаев развивается на фоне органического поражения структур головного мозга. У небольшого числа больных установить причину не удается.
На патологию приходится 2-3% от всех случаев детской эпилепсии и около 25% от младенческой. Средняя распространенность среди новорожденных — 3-5 больных на 10 тыс. детей. Болезнь в 60% случаев встречается у мальчиков. Чаще всего первые признаки возникают до года. Пик манифестации — 4-6 месяц жизни. Картина заболевания меняется при взрослении. К возрасту 3 лет клинические проявления проходят, либо у пациента развивается эпилепсия другой формы.
Причины развития
Синдром Веста развивается на фоне первичного поражения головного мозга: энцефалит, гипоксические повреждения нервной системы, внутричерепная родовая травма, асфиксия новорожденного, внутриутробные инфекции и пр. Симптомы болезни отмечаются у новорожденных с аномалиями строения ЦНС: увеличением больших полушарий, недоразвитием мозолистого тела и т.п. Возможно появление эпилептических приступов на фоне факоматозов — нейрофиброматоза и туберозного склероза, и генетических болезней (инфантильные спазмы при фенилкетонурии, синдроме Дауна).
У 10% больных развитие патологии имеет идиопатический характер. Конкретная причина не может быть выявлена. Врачи считают, что в этих случаях болезнь носит наследственный характер и связана с генетическими аномалиями.
Механизм развития инфантильных спазмов продолжает изучаться. Основная гипотеза — нарушение работы нейронов, вырабатывающих серотонин. Это приводит к дисбалансу нейромедиаторов и может провоцировать развитие судорожных приступов. Характер подобных нарушений не объяснен. Другая теория связывает возникновение синдрома Веста с иммунологическими изменениями в организме, а именно с увеличением количества активных В-лимфоцитов. Гипотеза согласуется с эффективностью применения иммуносупрессивных препаратов в лечении.
Самолечение и использование народных методов терапии при судорожных синдромах недопустимо. Это может стать причиной прогрессирования патологии и развития осложнений.
Клинические проявления
Признаки заболевания появляются в первый год жизни. В редких случаях возможна манифестация патологии до 4-летнего возраста. Основные симптомы — отставание в психомоторном развитии и мышечные спазмы.
У большинства детей сначала возникают признаки нейропсихического развития, на фоне которых появляются судорожные приступы. У 30% больных мышечные спазмы являются первыми проявлениями. Родители отмечают, что у детей не развиты хватательный рефлекс, слежение за предметами, а также способность фиксировать свой взгляд. При внешнем осмотре заметна общая мышечная гипотония.
Спазмы мышц возникают внезапно. Они проявляются симметрично и проходят в течение 1 минуты. Для синдрома Веста характерно их серийное возникновение. Интервал между отдельными сокращениями различен. Количество приступов в сутки — от 1 до нескольких сотен. Чаще всего мышечные спазмы возникают утром после пробуждения или в вечернее время перед засыпанием. Громкий звук и тактильные прикосновения могут провоцировать спастические приступы.
Мышечные спазмы могут носить изолированный или генерализованный характер. В первом случае сокращения наблюдаются в определенной группе мышц. Их разделяют на несколько видов:
- разгибательные (экстензорные);
- сгибательные (флексорные);
- смешанные.
Смешанные мышечные спазмы встречаются наиболее часто. Реже наблюдаются изолированные сгибательные или разгибательные сокращения. Характер приступа определяется положением тела в момент его начала.
Бессудорожные приступы встречаются редко. Они протекают по типу абсанса — ребенок замирает на одном месте на несколько секунд, после чего возвращается к прерванной деятельности. Инфантильные спазмы могут затрагивать только мышцы глазных яблок. Клиническая картина заболевания у всех детей имеет свои особенности.
Диагностические мероприятия

Обследование у детей носит комплексный характер
Триада признаков при синдроме Веста:
- гипсаритмический ЭЭГ-паттерн;
- отставание в психомоторном развитии;
- локальные мышечные спазмы.
В диагностике важную роль играет ранний возраст больного, что позволяет отличать заболевание от синдрома Драве и других болезней с судорожными проявлениями. Если болезнь возникает в возрасте 3-4 лет, то постановка диагноза затруднена. В процессе обследования ребенка осматривают педиатр, генетик, детский невролог и эпилептолог — они проводят дифференциальную диагностику.
Всем детям проводят компьютерную томографию головного мозга. Так как синдром Веста носит вторичный характер, то при нейровизуализации определяются очаговые и диффузные очаги поражения структур центральной нервной системы. Дополнительно возможно проведение МРТ или позитронно-эмиссионной томографии головного мозга.
Подходы к лечению
Длительное время заболевание было неизлечимо. В конце 60-х годов прошлого века был показан положительный эффект от приема препаратов адренокортикотропного гормона. Его применение и использование глюкокортикостероидов позволяет сократить число или полностью избавиться от спастических приступов, а также нормализовать картину мозговой активности на ЭЭГ. Схема лечения и дозировка лекарственных средств подбирается индивидуально. Средняя продолжительность терапии — 3-6 недель.
При выявлении первичного туберозного склероза назначаются медикаменты с вигабатрином. Это вещество обладающее сильным противоэпилептическим эффектом. Лечение им быстро приводит к снижению частоты спазмов. Среди других антиконвульсантов возможно назначение препаратов на основе вальпроевой кислоты. Положительный эффект имеют большие дозы витамина В6 при их введении на начальной стадии развития патологии.
Если консервативная терапия не дает эффекта, то проводятся нейрохирургические вмешательства. Конкретный вид операции определяет нейрохирург после осмотра ребенка.
Лекарственные средства имеют показания и противопоказания к применению. Их назначает только лечащий врач.
Прогноз
У 40-50% больных заболевание проходит при достижении 3-летнего возраста. В оставшихся случаях патология переходит в другой тип эпилептической патологии, например в синдром Леннокса-Гасто. В случае одновременного наличия синдрома Дауна болезнь плохо поддается консервативной терапии. Родителям важно понимать, что несмотря на устранение мышечных спазмов, задержка в психомоторном развитии сохраняется. Это приводит к поведенческим, когнитивным дефектам, а также к трудностям в обучении.
Говоря о том, сколько живут больные, врачи основывают прогноз на основании сопутствующих заболеваний. При наличии дефектов в строении головного мозга летальность достигает 25-30%. В других случаях уровень смертности не превышает 1%.
Синдром Веста - заболевание, имеющее благоприятный прогноз. Своевременное выявление болезни и комплексная терапия позволяют избавиться от спастических симптомов. Отставание в психомоторном развитии при этом сохраняется и требует длительной работы с ребенком для улучшения его адаптации. Диагностикой и терапией занимается врач-педиатр совместно с другими специалистами: неврологом и эпилептологом.
Первооткрыватель этой болезни нашел ее у своего сына, ее патогенез изучают 170 лет, а для лечения используют гормоны
Синдром Веста — младенческая эпилепсия, описанная впервые еще в 19 веке. До открытия влияния адренокортикотропного гормона (АКТГ) на течение этого заболевание оно считалось неизлечимым.
История
Итак, что это за болезнь? Синдром Веста (СВ) — это эпилептическая энцефалопатия у детей, проявляющаяся триадой:
- Инфантильные спазмы (ИС). Это короткие сильные сокращения мышц, соединяющих голову с позвоночником, и мышц, расположенных вдоль позвоночника (сгибательные, разгибательные или смешанные).
- Гипсаритмия — межприступные изменения на ЭЭГ.
- Прогрессирующее нарушение когнитивных, поведенческих и неврологических функций.
СВ встречается в 2–6 случаях на 10 000 новорожденных и составляет до 9 % эпилептических синдромов раннего детского возраста. От синдрома Веста чаще страдают мальчики — до 60 % от общего числа больных.
Формы
Официально СВ разделяют на симптоматическую (до 85 %), а также криптогенную и идиопатическую формы (вместе до 20 %). Но с клинической точки зрения у заболевания только 2 формы, так как различия между криптогенной и идиопатической формами практически отсутствуют. К симптоматической форме синдрома Веста относят случаи заболевания на фоне уже имеющейся патологии головного мозга или нарушений развития. У половины детей с симптоматической формой в анамнезе было осложненное течение внутриутробного периода: инфекции, метаболические расстройства, генетические и хромосомные дефекты (синдром Дауна и др.), а также нарушение внутриматочного кровообращения у матери. Реже наблюдается патология родового периода. Это гипоксически-ишемическое поражение мозга, травмы и другие осложнения в родах. К постнатальным причинам СВ относятся инфекции, травмы, гипоксически-ишемические инсульты и опухоли.
Криптогенную, или идиопатическую, форму заболевания диагностируют у детей с эпилепсией синдрома Веста без видимых причин, с нормальным психомоторным развитием и без повреждения головного мозга до возникновения заболевания. Это более благоприятная форма СВ.
Патогенез синдрома Веста в настоящее время неизвестен. У пациентов укорочена фаза REM-сна (фаза быстрого движения глаз), во время которой происходит нормализация ЭЭГ и снижение частоты спазмов. В связи с этим есть версия, что при СВ в стволе головного мозга имеет место дисфункция серотонинергических нейронов, участвующих в формировании циклов сна. Существуют и другие гипотезы, подразумевающие генетические и иммунные нарушения у маленьких пациентов.
Клиническая картина
Лечение
Важнейшая задача терапии — полное прекращение или снижение частоты приступов и подавление гипсаритмии, которая делает невозможным нормальное развитие ребенка. Противоэпилептические средства в этом случае малоэффективны. Так возможно ли излечение от синдрома Веста?
В 1958 году в Европейском журнале о неврологии (European Journal of Neurology) была опубликована важнейшая работа по эпилепсии и эффективности введения кортикотропина при инфантильных спазмах (авторы Л. Сорель и A. A. Дюшан-Бойоль). АКТГ помогал в 50–90 % случаев, причем лечению лучше поддавалась криптогенная форма, чем симптоматическая. В большом финском исследовании 1980 года летальные осложнения при терапии кортикотропином достигали 5 %, а частота серьезных побочных эффектов составила 37 %. Высокий риск осложнений и низкая эффективность кортикотропина при симптоматической форме СВ привели к необходимости дальнейшего поиска препаратов для купирования спазмов.
Сейчас используются и другие гормональные средства — преднизолон, дексаметазон и тетракозактид. Последний препарат — это синтетический полипептид, обладающий свойствами эндогенного кортикотропина и дающий меньше осложнений, чем сам кортикотропин. В течение последних 20 лет зарекомендовал себя противоэпилептический препарат вигабатрин. Восприимчивость к терапии составляет 23–68 %. До сих пор не определены оптимальные дозы и продолжительность лечения ни для вигабатрина, ни для кортикотропина и тетракозактида.
Кроме того, при лечении синдрома Веста назначают вальпроаты и бензодиазепины. Однако полное исчезновение инфантильных спазмов на фоне приема этих препаратов наступает позже, чем при лечении стероидами и вигабатрином. При локализованном очаге эпилептоидной активности возможно хирургическое лечение, однако эффективно оно далеко не во всех случаях.
Динамика обязательно оценивается ЭЭГ-мониторингом, поскольку на фоне терапии спазмы могут перейти в субклинические, которые трудно распознать без ЭЭГ. В ремиссии (месяц без приступов) гипсаритмия может полностью исчезнуть, сменяясь нормальным вариантом ЭЭГ. Но в 23–50 % случаев синдрома Веста прогноз не очень хороший — заболевание трансформируется в другие формы эпилепсии, которые иногда могут проявить себя только в пубертатном периоде.
Прогноз
Синдром Веста — серийные спастические сокращения в отдельных мышечных группах или генерализованного характера, протекающие на фоне задержки нейропсихического развития и сопровождающиеся гипсаритмическим ЭЭГ-паттерном. Манифестирует в возрасте до 4-х лет, преимущественно на 1-ом году жизни. В большинстве случаев имеет симптоматический характер. Диагностика синдрома основана на клинических данных и результатах ЭЭГ. Для выявления основной патологии необходимы КТ или МРТ, ПЭТ головного мозга, консультация генетика, нейрохирурга. Лечение возможно противоэпилептическими препаратами, стероидами (АКТГ, преднизолон), вигабатрином. По показаниям решается вопрос о хирургическом лечении (каллозотомия, удаление патологического очага).
- Причины синдрома Веста
- Симптомы синдрома Веста
- Диагностика синдрома Веста
- Лечение синдрома Веста
- Прогноз синдрома Веста
- Цены на лечение
Общие сведения
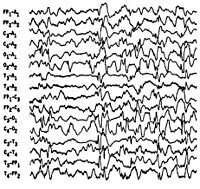
Синдром Веста носит название по имени врача, наблюдавшего его проявления у своего ребенка и впервые описавшего его в 1841 г. В связи с манифестацией синдрома в раннем возрасте и протеканием судорог по типу серии отдельных спазмов, пароксизмы, характеризующие синдром Веста, получили название инфантильные спазмы. Первоначально заболевание относили к генерализованной эпилепсии. В 1952 г. был изучен специфический гипсаритмический ЭЭГ-паттерн, патогномоничный для этой формы эпилепсии и характеризующийся медленноволновой асинхронной активностью с беспорядочными спайками высокой амплитуды. В 1964 г. специалистами в области неврологии синдром Веста был выделен в качестве отдельной нозологии.
Внедрение в неврологическую практику нейровизуализации позволило определить наличие у пациентов очаговых поражений вещества мозга. Это заставило неврологов пересмотреть свои взгляды на синдром Веста как на генерализованную эпилепсию и отнести его в ряд эпилептических энцефалопатий. В 1984 г. был выявлена эволюция эпилептической формы энцефалопатии от ее раннего варианта в синдром Веста, а с течением времени в синдром Леннокса-Гасто.
В настоящее время синдром Веста занимает около 2% от всех случаев эпилепсии у детей и примерно четверть младенческой эпилепсии. Распространенность составляет, по различным источникам, от 2 до 4,5 случаев на 10 тыс. новорожденных. Несколько чаще заболевают мальчики (60%). 90% случаев манифестации синдрома приходится на 1-й год жизни, с пиком в возрасте от 4 до 6 мес. Как правило, к возрасту 3-х лет мышечные спазмы проходят или трансформируются в иные формы эпилепсии.
Причины синдрома Веста
В подавляющем большинстве случаев синдром Веста носит симптоматический характер. Он может возникать вследствие перенесенных внутриутробных инфекций (цитомегалии, герпетической инфекции), постнатального энцефалита, гипоксии плода, преждевременных родов, внутричерепной родовой травмы, асфиксии новорожденного, постнатальной ишемии вследствие позднего пережатия пуповины. Синдром Веста может являться следствием аномалий строения головного мозга: септальной дисплазии, гемимегалоэнцефалии, агенезии мозолистого тела и пр. В ряде случаев инфантильные спазмы выступают симптомом факоматозов (синдрома недержания пигмента, туберозного склероза, нейрофиброматоза), точечных генных мутаций или хромосомных аберраций (в т. ч. синдрома Дауна). В литературе упоминаются случаи фенилкетонурии с инфантильными спазмами.
В 9-15% синдром Веста является идиопатическим или криптогенным, т. е. его первопричина не установлена или не очевидна. Зачастую при этом прослеживается наличие случаев фибрильных судорог или эпиприступов в семейном анамнезе больного ребенка, т. е. имеет место наследственная предрасположенность. Ряд исследователей указывают, что фактором, провоцирующим синдром Веста, может выступать вакцинация, в частности введение АКДС. Это может быть связано с совпадением сроков вакцинации и возраста типичного дебюта синдрома. Однако достоверные данные, подтверждающие провоцирующую роль вакцин, пока не получены.
Симптомы синдрома Веста
Как правило, симптом Веста дебютирует на первом году жизни. В отдельных случаях его манифестация происходит в более старшем возрасте, однако не позже 4-х лет. Основу клиники составляют серийные мышечные спазмы и нарушение психомоторного развития. Первые пароксизмы зачастую появляются на фоне уже существующей задержки психомоторного развития (ЗПР), но в 1/3 случаев возникают у первично здоровых детей. Отклонения в нейропсихологическом развитии наиболее часто проявляются снижением и выпадением хватательного рефлекса, аксональной гипотонией. Возможно отсутствие слежения глазами за предметами и расстройство фиксации взора, что является прогностически неблагоприятным критерием.
Мышечные спазмы носят внезапный симметричный и кратковременный характер. Типична их серийность, при этом интервал между следующими друг за другом спазмами длится не менее 1 минуты. Обычно наблюдается возрастание интенсивности спазмов в начале пароксизма и ее спад в конце. Число спазмов, происходящих за сутки, варьирует от единиц до сотен. Наиболее часто возникновение инфантильных спазмов происходит в период засыпания или сразу после сна. Провоцировать пароксизм способны резкие громкие звуки и тактильная стимуляция.
Семиотика пароксизмов, которыми сопровождается синдром Веста, зависит от того, какая мышечная группа сокращается — экстензорная (разгибательная) или флексорная (сгибательная). По этому признаку спазмы классифицируют на экстензорные, флексорные и смешанные. Чаще всего наблюдаются смешанные спазмы, затем сгибательные, наиболее редко — разгибательные. В большинстве случаев у одного ребенка наблюдаются спазмы нескольких видов и то, какой именно спазм будет преобладать, зависит от положения тела в момент начала пароксизма.
Наряду с серийными спазмами, синдром Веста может сопровождаться бессудорожными приступами, проявляющимися внезапной остановкой двигательной активности. Иногда отмечаются пароксизмы, ограниченные подергиванием глазных яблок. Возможно нарушение дыхания вследствие спазма дыхательной мускулатуры. В некоторых случаях имеют место асимметричные спазмы, проявляющиеся отведением головы и глаз в сторону. Могут встречаться и другие виды эпиприступов: фокальные и клонические. Они комбинируются со спазмами или имеют самостоятельный характер.
Диагностика синдрома Веста
Синдром Веста диагностируется по основной триаде признаков: приступы кластерных мышечных спазмов, задержка психомоторного развития и гипсаритмический ЭЭГ-паттерн. Имеют значение возраст манифестации спазмов и их связь со сном. Трудности диагностики возникают при позднем дебюте синдрома. В ходе диагностики ребенок консультируется педиатром, детским неврологом, эпилептологом, генетиком. Дифференцировать синдром Веста следует с доброкачественным младенческим миоклонусом, доброкачественной роландической эпилепсией, младенческой миоклонической эпилепсией, синдромом Сандифера (наклон головы по типу кривошеи, гастроэзофагальный рефлюкс, эпизоды опистотонуса, которые могут быть приняты за спазмы).
КТ головного мозга у имеющих синдром Веста детей может выявлять диффузные либо очаговые изменения церебральных структур, но может быть в пределах нормы. В диагностике локальных поражений более чувствительным методом является МРТ головного мозга. Для выявления участков гипометаболизма мозговых тканей в некоторых случаях возможно проведение ПЭТ головного мозга.
Лечение синдрома Веста
Синдром Веста считался резистентным к проводимой терапии вплоть до открытия в 1958 г. влияния на приступы препаратов АКТГ. Терапия АКТГ и преднизолоном приводит к значительному улучшению или полному прекращению инфантильных спазмов, что сопровождается исчезновением гипсаритмического ЭЭГ-паттерна. До сих пор среди неврологов нет однозначных решений касательно доз и длительности стероидной терапии. Исследования показали, что в 90% случаев терапевтический успех достигался при применении больших дозировок АКТГ. Сроки терапии могут варьировать в пределах 2-6 недель.
Новый этап в лечении инфантильных спазмов начался в 1990-1992 гг. после обнаружения положительного терапевтического эффекта вигабатрина. Однако преимущество лечения вигабатрином пока доказано лишь для больных туберозным склерозом. В остальных случаях исследования показали большую эффективность стероидов. С другой стороны стероидная терапия имеет худшую, в сравнении с вигабатрином, переносимость и более высокий процент рецидивов.
Из антиконвульсантов эффективность показана лишь у нитразепама и вальпроевой кислоты. У отдельных пациентов описан лечебный эффект больших доз витамина В6, который отмечался в первые недели терапии. При инфантильных спазмах, резистентных к проводимой терапии, с подтвержденным на томографии наличием патологического очага показана консультация нейрохирурга для решения вопроса о резекции очага. Если подобная операция невозможна, то при наличии дроп-атак проводится тотальная каллозотомия (пересечение мозолистого тела).
Прогноз синдрома Веста
Обычно к 3-летнему возрасту наблюдается регресс и исчезновение инфантильных спазмов. Но примерно в 55-60% случаев они трансформируются в другую форму эпилепсии, чаще всего в синдром Леннокса-Гасто. Фармакорезистентность часто констатируется при инфантильных спазмах, сопровождающих синдром Дауна. Даже при успешном купировании пароксизмов синдром Веста имеет неудовлетворительный прогноз в плане психомоторного развития ребенка. Возможны когнитивные и поведенческие нарушения, ДЦП, аутизм, трудности в обучении. Остаточный психомоторный дефицит не наблюдается только в 5-12% случаев. ЗПР отмечается у 70-78% детей, двигательные расстройства — у 50%. Серьезный прогноз имеет синдром Веста, обусловленный аномалиями или дегенеративными изменениями головного мозга. При этом летальность может достигать 25%.
Более благоприятный прогноз имеют криптогенный и идиопатический синдром Веста при отсутствии ЗПР до появления спазмов. В этой группе больных остаточный интеллектуальный или неврологический дефицит отсутствует у 37-44% детей. Неблагоприятно отражается на прогнозе болезни откладывание начала лечения. Прогностическая оценка затрудняется тем, что отдаленные последствия также зависят от основной патологии, на фоне которой возникает симптоматический синдром Веста.
Читайте также: