
Протоонкогены участвуют в развитии опухолей
Определение
В клетке с нормальной регуляцией (без рака) гены продуцируют белки, которые обеспечивают регулируемое деление клеток. Рак — это заболевание, вызванное клетками, которые потеряли способность контролировать свое регулирование. Аномальные белки, разрешающие нерегулируемое раковое состояние, продуцируются генами, известными как онкогены. Нормальный ген, из которого развился онкоген, называется протоонкогеном.
Описание
Слово онкоген происходит от греческого термина oncos, что означает опухоль. Онкогены были первоначально обнаружены у некоторых видов животных вирусов, которые были способны индуцировать опухоли у инфицированных животных. Эти вирусные онкогены, называемые v-onc, позднее были обнаружены в опухолях человека, хотя большинство раковых заболеваний человека, по-видимому, не вызвано вирусами. С момента своего первоначального открытия были найдены сотни онкогенов, но известно лишь небольшое число из них, которые влияют на людей. Хотя разные онкогены имеют разные функции, все они каким-то образом вовлечены в процесс трансформации (изменения) нормальных клеток в раковые клетки.
Преобразование нормальных клеток в раковые клетки
Процесс, посредством которого нормальные клетки трансформируются в раковые, представляет собой сложный многоэтапный процесс, включающий распад нормального клеточного цикла. Обычно соматическая клетка проходит цикл роста, в котором она продуцирует новые клетки. Двумя основными этапами этого цикла являются мейоз (генетический материал дублируется) и митоз (клетка делит на две другие идентичные клетки). Процесс деления клеток необходим для роста тканей и органов тела и для замены поврежденных клеток. Нормальные клетки имеют ограниченный срок службы и проходят цикл ограниченное количество раз. Рак в одном направлении обусловлен дерегуляцией тех генов, которые связаны с контролем клеточного цикла. Если онкоген присутствует в клетке кожи, у пациента будет рак кожи; в молочной клетке, her2 молочной железы, и так далее. Клетки, которые теряют контроль над их клеточным циклом и уходят из-под контроля, называются раковыми клетками. Раковые клетки подвергаются многим клеточным делениям быстрее, чем обычные клетки, и не имеют ограниченного срока службы. Это позволяет им в конечном итоге подавить тело большим количеством аномальных клеток и повлиять на функционирование нормальных клеток. Клетка становится раковой только после того, как изменения происходят в ряде генов, которые участвуют в регуляции клеточного цикла. Изменение регуляторного гена может заставить его прекратить продуцировать нормальный регуляторный белок или может вызвать аномальный белок, который не регулирует клетку нормальным образом. Когда изменения происходят в одном регуляторном гене, это часто вызывает изменения в других регуляторных генах. Раки в различных типах клеток могут быть вызваны изменениями в различных типах регуляторных генов. Протоонкогены и гены-супрессоры опухолей являются двумя наиболее распространенными генами, участвующими в регуляции клеточного цикла.
Протоонкогены и гены-супрессоры опухолей имеют различные функции в клеточном цикле. Гены опухолевых супрессоров продуцируют белки, которые участвуют в предотвращении неконтролируемого роста и деления клеток. Прото-онкогены производят белки, которые в основном участвуют в стимулировании роста и деления клеток контролируемым образом. Каждый протоонкоген продуцирует другой белок, который играет уникальную роль в регулировании клеточных циклов конкретных типов клеток. Изменение только одного протоонкогена пары превращает его в онкоген. Онкоген производит ненормальный белок, который каким-то образом участвует в стимулировании неконтролируемого роста клеток. Онкоген действует аутосомно-доминантным образом, так как только один протоонкоген пары нуждается в изменении в формировании онкогена.
Открытие онкогенов в нормальных клетках животных и человека ставит вопрос о возможной их роли в развитии невирусных опухолей, т. е. вопрос о едином патогенетическом механизме возникновения опухолей активацией онкогенов канцерогенными агентами различной природы (химическими, физическими, биологическими). Обсуждается вопрос о том, каким образом гены, программирующие образование злокачественной опухоли, не были элиминированы в процессе эволюции и сохранились в качестве нормальных составных частей клеточного генома.
Предполагают, что онкогенность является побочным результатом действия этих генов и проявляется лишь в исключительных случаях, основной же их функцией является участие в процессах нормальной пролиферации и диффереицировки клеток. Так, показано, что онкоген c-ras экспрессируется при регенерации печени у крыс и возвращается к исходному уровню по завершении регенерации. Особо важную роль онкогены, по-видимому, играют в процессе эмбриогенеза.
В настоящее время идентифицировано около 40 онкогенов, определяющих опухолеродную активность вирусов птиц, грызунов и обезьян. Установлена локализация клеточных онкогенов в хромосомах человека, при этом выяснилось, что они локализуются не только именно в тех хромосомах, в которых обнаружены специфические перестройки при злокачественных новообразованиях, но и в тех местах, которые нарушаются при этих перестройках. Так, при хроническом миелоидиом лейкозе онкоген с-аЫ переносится при траислокации с 9-й хромосомы на 22-ю, а при лимфоме Беркитта ген с-myc — с 8-й на 14-ю
Смысл этих специфических перестроек заключается в том, что онкоген переносится в активные участки генома, что сопровождается активацией онкогена В действительности процесс малигнизации значительно сложнее и требует, как считают, активации нескольких онкогенов Ныне установлена локализация на хромосомах человека более 40 онкогенов, в нх числе упомянутые выше онкогены с-аЫ и с-myc (названия генов составлены из трех латинских букв, взятых из названий соответствующих вирусов abi — вирус лейкоза мышей Абельсона, myc — вирус птичьего миелоцитоматоза, srс — вирус саркомы Рауса, вирус мышиной саркомы Молони и т. д.)
Активированные трансформирующие гены (онкогены) выявлены в клетках таких опухолей человека, как рак мочевого пузыря, легкого, молочной железы, толстой кишки, поджелудочной железы, нейробластома, В- и Т-клеточная лимфома, фибро- н рабдомиосаркома и др.
Важным открытием было обнаружение сходства продукта экспрессии онкогена с нормальным белковым фактором роста кровяных пластинок. Это позволило предположить, что злокачественная трансформация клеток онкогеном может осуществляться путем избыточного производства продукта, в норме стимулирующего рост. Если, как указывает И Ф Сейц (1984), такая закономерность будет установлена, то причину злокачественной трансформации нужно будет искать не в качественных, а в количественных изменениях механизмов, регулирующих рост на нормальной физиологической основе.
Эта область исследовании развивается очень бурно, и возможно, что будут получены ответы иа такие вопросы, как место онкогенов в многостадийном канцерогенезе (является ли активация онкогена лишь пусковым механизмом, а остальные стадии процесса развиваются автономно, или для каждой стадии требуется активация специального онкогена); степень специфичности онкогенов (вызывается ли каждый тип новообразования своим собственным онкогеном или один онкоген или какая-то комбинация онкогенов могут быть ответственны за возникновение разных опухолей). Решение этих вопросов имеет прямое отношение к практике.
Как видно из приведенных данных, прямая этиологическая роль вирусов в возникновении злокачественных опухолей человека доказана пока лишь в единичных случаях (это Т-клеточный лейкоз взрослых и, вероятно, африканская лимфома Беркитта). В свое время выдвигались концепции о едином механизме канцерогенеза, осуществляемом за счет гипотетических провирусов или протовирусов В настоящее время вирусный канцерогенез рассматривается лишь как частный случай канцерогенеза, а общим звеном в возникновении опухолей любой этиологии считается активация, превращение собственных клеточных генов (протоонкогенов) в онкогены.
Протоонкоген MYC присутствует фактически во всех клетках эукариотов и принадлежит к группе немедленно реагирующих генов, индуцируемых после получения сигнала к делению покоящейся клеткой. После краткосрочного повышения МУС экспрессия мРНК снижается до исходного уровня.
Молекулярные механизмы функционирования MYC в делящихся клетках остаются до конца не понятыми. Полагают, что участие MYC в канцерогенезе, как и других факторов транскрипции, связано с активацией генов, участвующих в пролиферации. Действительно, некоторые из генов-мишеней MYC — орнитиндекарбоксилаза и циклин D2 — задействованы в клеточной пролиферации.
Однако диапазон активирующего влияния MYC гораздо шире и включает ацетилирование гистонов, снижение адгезивности клеток, увеличение клеточной подвижности, увеличение синтеза белка, снижение протеазной активности и другие изменения метаболизма, обеспечивающие высокий уровень деления клеток. Геномная карта связывающих сайтов MYC содержит тысячи различных участков и эквивалентное количество генов, которые могут регулироваться.
Однако в связи с тем, что генами-мишенями MYC при разных злокачественных опухолях являются разные гены, универсальной концепции участия MYC в канцерогенезе пока нет. Небезынтересно высказанное предположение, что MYC взаимодействует с членами системы ДНК, запускающими процессы репликации, поэтому усиление экспрессии МУС вызывает активацию большего количества генов, участвующих в репликации, чем это необходимо для нормального деления клеток, или дает возможность клеткам уклоняться от точек контроля, что приводит к повреждению генома и накоплению мутаций.
Итак, MYC находится в числе небольшого количества факторов, способных вызывать перепрограммирование соматических клеток в плюрипотентные стволовые клетки; MYC также может усиливать восстановление и дифференцировку клеток.
С одной стороны, активация MYC приводит к пролиферации, а с другой — к апоптозу при отсутствии факторов роста, что было показано на клеточных культурах. Протоонкоген MYC имеет отдельные домены, активирующие как рост, так и апоптоз клеток, однако остается недоказанным, может ли возникать MYC-индуцированный апоптоз in vivo.
В опухолях обнаруживаются персистенция или чрезмерная экспрессия белка MYC в отличие от регулируемой экспрессии при нормальной клеточной пролиферации. Дисрегуляция гена MYC в результате транслокации наблюдается в лимфоме Беркитта, опухоли из В-клеток. MYC амплифицирован в некоторых случаях карцином молочной железы, толстой кишки, легкого и многих других карциномах. Родственные гены N-MYC и L-MYC амплифицированы в нейробластомах и мелкоклеточном раке легкого соответственно.
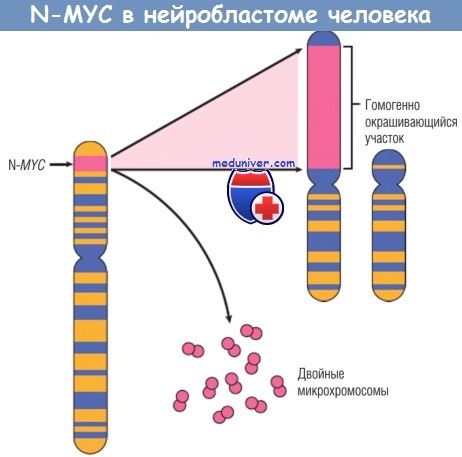
Амплификация N-MYC в нейробластоме человека.
Ген U-MYC в норме локализуется на хромосоме 2р, при амплификации выявляется как добавочные двойные микрохромосомы или гомогенно окрашивающийся участок хромосомы, с которой он интегрировался.
Интеграция обычно происходит с аутосомами 4, 9 или 13.
- 1469
- 1,1
- 1
- 5
Малигнизация — один из самых загадочных процессов. Что же на самом деле направляет клетку на тернистый путь перерождения?
![]()
Анна Батуева




Спонсором приза зрительских симпатий выступила компания BioVitrum.
Гончие еще играют во дворе, но дичи не уйти,
как ни мчится она уже сейчас по лесам.
Франц Кафка
Под малигнизацией понимают приобретение здоровыми клетками черт злокачественности, которые мы подробно рассмотрим ниже. Процесс злокачественного изменения можно уподобить дичи из цитаты Кафки, ведь клетка, однажды встав на этот путь, не сможет вернуться и получить свое клеточное здоровье обратно. Важную роль в понимании основ перерождения клеток и их дальнейшего функционирования сыграла медицина, а следом за ней и молекулярная биология. Но начнем с истоков истории рака.
Часть 1. Биография рака
Первые упоминания о раке встречаются в папирусе Эдвина Смита, датируемом 16 веком до нашей эры [1]. Там же отмечается, что данное заболевание не поддается лечению.
Во времена Гиппократа, около 400 года до нашей эры, появилось специальное обозначение рака — karkinos. Разросшаяся опухоль напомнила Гиппократу краба, окутывающего все вокруг клешнями. Современное название онкологии произошло от греческого слова onkos, которое греки использовали для описания опухолей. Однако врачи того времени не различали доброкачественные и злокачественные новообразования, и karkinos Гиппократа не имеет ничего общего с истинным раком.
Гиппократ выдвинул гуморальную теорию, суть которой состояла в том, что каждый недуг является следствием переизбытка одного из четырех гуморов: крови, слизи, желтой желчи и черной желчи.
Первая половина XX века породила еще одну теорию канцерогенеза, недалеко ушедшую от истины. В 1911 году Пейтон Раус, работая в Рокфеллеровском университете в Нью-Йорке, открыл вирус, способный вызывать опухоли у кур. Ученые по всему свету бросились искать вирусы, ответственные за рак именно у человека, однако ничего не могли найти. В 1974 году в Medical World News вирус рака у человека ставили в один ряд с НЛО, снежным человеком и лохнесским чудовищем. Вирус СВ-40 и вирус папилломы человека, вызывающие рак у людей, были открыты в 1960 и 1983 годах соответственно.
В 1970 году генетик Говард Темин, работавший в лаборатории Макардла в Висконсине и изучавший вирус саркомы Рауса (ВСР, или VSR), представил свою работу на Десятом Международном онкологическом конгрессе. Он открыл у ВСР обратную транскрипцию — синтез ДНК по РНК — и положил начало изучениям ретровирусов. Позднее он отказался от вирусной теории канцерогенеза, а в 1979 году ученые Майкл Бишоп и Харолд Вармус открыли первый протоонкоген — src (сарк), содержащийся в ВСР. Это положило начало новому этапу в истории онкологии, люди наконец-то поняли, как запускается процесс канцерогенеза. Но этого бы не произошло без изучения раковой клетки и ее странной физиологии.
Часть 2. Что заставляет клетку измениться?
В этой главе мы разберем причины злокачественного перерождения клетки. Первым толчком к началу этого изменения является мутация в ДНК.
Важными факторами, вызывающими мутации и провоцирующими раковое перерождение, являются ионизирующее излучение, воздействие ультрафиолетовых лучей, влияние цитотоксических веществ, повреждающих ДНК (к ним относятся наркотические вещества и некоторые лекарственные препараты — например, цисплатин, повреждающий структуру двойной спирали) и органические яды.
Но не всякие повреждения ДНК обязательно приведут к появлению раковой клетки, а лишь те, что затронут определенные гены. Наиболее важную роль в канцерогенезе играют три группы генов: протоонкогены, онкогены и гены — супрессоры опухолей.
Основные изменения, происходящие с протоонкогенами:
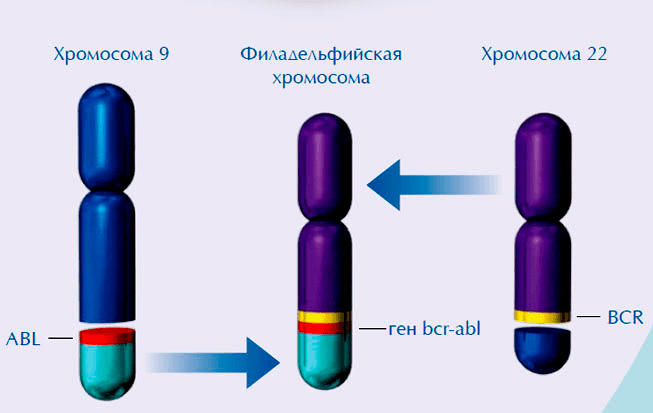
Рисунок 1. Химерный ген BCR-ABL образуется при слиянии участка 9 хромосомы, несущей ген ABL, с участком 22 хромосомы, несущей ген BCR
В некоторых случаях канцерогенез запускается вирусами. Онкогены в геноме вирусов являются ранее захваченными в клетках-хозяевах нормальными генами, которые со временем превратились в злокачественные. Когда такие онкогенные вирусы попадают в клетку, начинается считывание информации с вирусной ДНК или РНК, в цитоплазме накапливаются онкогенные белки и начинается процесс перерождения.
Онкогены — это гены, активность которых стимулирует образование и развитие злокачественной опухоли. Как уже было упомянуто выше, первый вирусный онкоген был открыт в 1979 году.
Онкогены кодируют белки с различной структурой и функциями. К основным продуктам деятельности онкогенов относят:
- Факторы роста. Раковые клетки продуцируют белки, способные вызывать пролиферацию и дифференцировку клеток. Наиболее известным фактором роста является HER2, кодируемый геном ERBB2. Мутации и гиперэкспрессия этого гена обнаружены при раке молочной железы и ассоциированы с крайней агрессивностью опухоли. Суперэкспрессия гена приводит к запуску белковых каскадов, ответственных за клеточное деление. Постоянные сигналы к делению вызывают неконтролируемую пролиферативную активность клеток и их злокачественное перерождение.
- ГТФ-связывающие белки. Гуанозинтрифосфат-связывающие белки участвуют во многих клеточных процессах: передача сигналов, транспорт метаболитов внутри клетки и др. Первыми открытыми ГТФ-связывающими белками были белки семейства Ras — продукты онкогена RAS. При постоянном производстве они вызывают злокачественный рост. Наиболее изученный эффектор Ras — это RAF, который запускает белковый каскад MAPK, отвечающий за клеточное деление и пролиферацию [6].
- Мембранные рецепторы. В онкогенезе основную роль играют рецепторы с тирозинкиназной активностью. Они служат для связывания с ростовыми факторами. К ним относится рецептор эпидермального фактора роста, повышенный синтез которого приводит к перерождению клетки.
- Онкогенные протеинкиназы. Протеинкиназы — это группа ферментов, которые модифицируют белки путем фосфорилирования (присоединения остатка фосфорной кислоты). Протеинкиназы регулируют апоптоз, процессы роста и дифференцировки клеток. Нарушения в их работе приводят к сбою в клеточном цикле и, как следствие, к развитию рака. Например, протеинкиназа AKT1, ответственная за ингибирование апоптоза, при перепроизводстве способна вызывать перерождение клеток. Также, она связана с ростом сосудов в опухоли, что помогает раковым клеткам расселяться по организму и давать метастазы.
Все вышеперечисленные продукты онкогенов являются сигналами к запуску неконтролируемого клеточного деления. Внешние факторы больше не играют никакой роли в жизни клетки, потому что пролиферацию запускают внутренние сигнальные белки.
В здоровой клетке существуют защитные механизмы, следящие за процессами и регулирующие клеточный цикл. К таким механизмам относят деятельность белков — супрессоров опухолей: p21, p53, pRb, PTEN и др.
Белок p53 — наиболее изученный белок-супрессор. Он является продуктом гена TP53, мутации которого обнаруживаются в клетках многих опухолей [7]. p53 синтезируется во всех клетках организма, но активируется только при повреждениях ДНК. Этот белок способен остановить клеточный цикл и не допускать дальнейшее деление клетки, пока не произойдет репарация ДНК. При сильных повреждениях он также может запускать процесс апоптоза.
Одной из главных функций p53 является сохранение генетической идентичности всех клеток организма. При неправильной работе этого белка клетка получает возможность делиться даже при поврежденной ДНК, что увеличивает вероятность мутаций и накопления дефектных онкогенов. Важную роль в подавлении p53 играет белок MDM2, который в норме регулирует активность p53. Однако при повышенном синтезе он связывается с p53 и ингибирует его противоопухолевое действие.
Важными факторами канцерогенеза являются эпигенетические события. Эпигенетика изучает процессы, затрагивающие активность генов, но не изменяющие структуру ДНК. К ним относится изменение метилирования ДНК.
Метилирование — это присоединение метильной группы к нуклеотидам в особых, строго определенных участках генома, называемых CpG-островками. Такое изменение не влияет на структуру молекулы, однако может влиять на экспрессию отдельных генов. В частности, если в участке ДНК много метильных групп, то транскрипция этого участка прекращается.
Особенно активно метилирование проходит в эмбриональный период жизни, а у взрослого человека метилировано около 2% генома. В норме баланс между метилированием и деметилированием строго регулируется и соблюдается, однако в старости начинают преобладать процессы метилирования, что может в итоге привести к канцерогенезу. В процессе онкогенеза происходит гиперметилирование CpG-островков, что приводит к общей геномной нестабильности и накоплению еще большего количества мутаций. В большинстве случаев метилированные участки являются промоторами и влияют на активацию или, наоборот, инактивацию генов, что с виду похоже на действие точечных мутаций.
Однако хотя нарушения в эпигенетической регуляции сопровождают развитие злокачественного перерождения, они, как правило, не являются его первопричиной, а лишь одним из сопутствующих факторов.
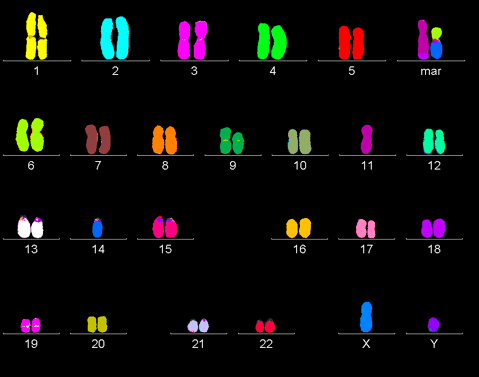
Рисунок 2. Раковый геном. В процессе жизнедеятельности раковая клетка накапливает огромное количество мутаций и нередко характиризуется полиплоидностью.
Часть 3. Физиологические последствия малигнизации
Основное последствие малигнизации — клеточное бессмертие. Оно может поддерживаться несколькими способами: активацией фермента теломеразы, блокировкой регуляторов митохондриального пути апоптоза и в некоторых случаях активацией механизма ALT (alternative lengthening of telomeres, альтернативного удлинения теломер [14]).
Впервые клеточное бессмертие раковых клеток было продемонстрировано в 1951 году на клеточной линии HeLa, взятой у Генриетты Лакс, вскоре скончавшейся от рака шейки матки (рис. 3) [9].

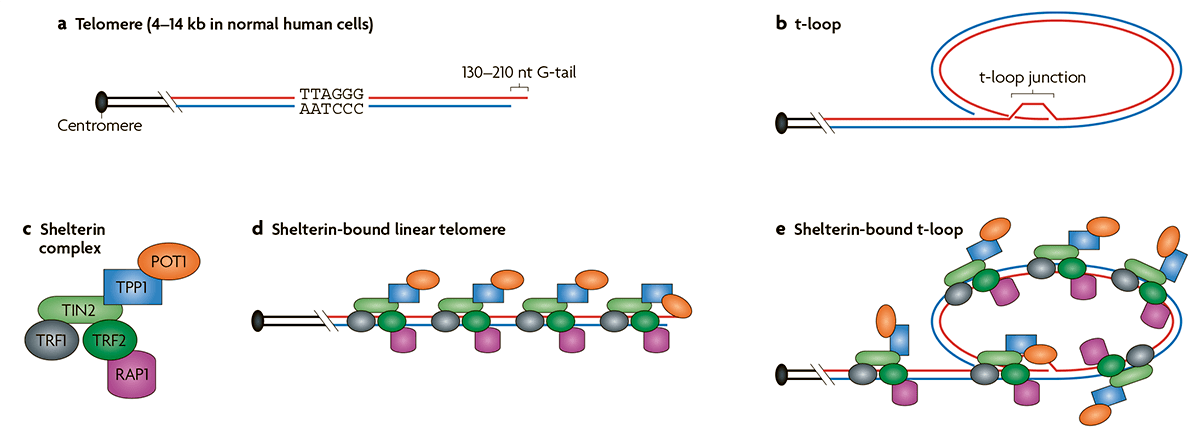
Как правило, малигнизация сопровождается активацией фермента теломеразы. На концах хромосом находятся короткие повторяющиеся участки ДНК, названные теломерами [10]. После каждого деления теломеры укорачиваются, что в итоге приводит к их полному исчезновению и невозможности продолжать деление. Количество возможных делений для клетки названо пределом Хейфлика. Действие теломеразы заключается в восстановлении теломер и превращении клетки в фактически бессмертную, позволяя ей делиться бесконечно долго. Существуют нормальные клетки, в которых также экспрессируется теломераза. Это клетки, которым надо часто делиться: половые, стволовые и клетки эпителия кишечника. Однако теломераза активна в подавляющем большинстве раковых клеток, что играет важную роль в их жизненном цикле.
С другой стороны, в некоторых злокачественных клетках, наравне с активной теломеразой, существует так называемое альтернативное удлинение теломер, или сокращенно ALT [11]. При ALT происходит гомологичная рекомбинация концевых участков хромосом (рис. 4). В норме рекомбинация происходит в процессе мейоза, однако раковые клетки научились достраивать теломеры, используя теломеры другой хромосомы как матрицу [12].
Важно отметить, что раковое бессмертие контролируется не только теломерами, но и ингибированием путей апоптоза, главным из которых является митохондриальный путь. В норме, из митохондрий в цитоплазму выходят митохондриальные белки и образуют апоптотический комплекс — апоптосому, которая и запускает апоптоз. При неправильной работе регуляторных белков, а к ним относятся белки семейства BCL-2, нарушается выход апоптотических белков, что приводит к сбою в процессе апоптоза. В раковых клетках обнаружены нарушения в работе белков BAX и BAK, а также экспрессия ингибиторов клеточной смерти.
Часть 4. Заключение

Существуют пять основных классов протоонкогенов / онкогенов:
- факторы роста,
- рецепторы фактора роста,
- преобразователи сигналов,
- факторы транскрипции,
- запрограммированные регуляторы смертности клеток.
Факторы роста
Некоторые протоонкогены продуцируют белки, называемые факторами роста, которые косвенно стимулируют рост клетки путем активации рецепторов на поверхности клетки. Различные факторы активируют различные рецепторы, обнаруженные на разных клетках организма. Мутации в протоонкогене фактора роста приводят к онкогенам, которые способствуют неконтролируемому росту клеток, для которых они имеют рецептор. Например, тромбоцитарный фактор роста (PDGF) представляет собой протоонкоген, который помогает стимулировать заживление ран, стимулируя рост клеток вокруг раны. PDGF можно мутировать в онкоген, называемый vsis (PDGFB), который часто присутствует в опухолях соединительной ткани.
Рецепторы
Факторы роста обнаруживаются на поверхности клеток. Факторы роста посылают сигналы в центр клетки (ядра) и стимулируют клетки, которые находятся в состоянии покоя, войти в клеточный цикл. Различные клетки имеют разные рецепторы факторов роста. Мутации в протоонкогене, которые являются рецепторами фактора роста, могут приводить к онкогенам, которые продуцируют рецепторы, которые не требуют факторов роста, чтобы стимулировать рост клеток. Чрезмерная стимуляция клеток для входа в клеточный цикл может приводить к развитию неконтролируемого роста клеток. Большинство рецепторов рецептора протоонкогена называются тирозинкиназами и очень вовлечены в контроль формы и роста клеток. Один пример тирозинкиназы называется GDFNR. РЕТ (перегруппированный во время трансфекции) онкоген является мутантной формой GDFNR и обычно встречается в раковых клетках щитовидной железы.
Сигнальные преобразователи
Сигнальные преобразователи — это белки, которые ретранслируют сигналы стимуляции клеточного цикла, от рецепторов фактора роста до белков в ядре клетки. Передача сигналов в ядро является ступенчатым процессом, который включает в себя большое количество протоонкогенов и часто называется каскадом передачи сигналов. Мутации в протоонкогене, участвующем в этом каскаде, могут вызывать нерегулируемую активность, что может привести к аномальной пролиферации клеток. Онкогены преобразователя сигналов являются крупнейшим классом онкогенов. Семейство RAS представляет собой группу из 50 связанных онкогенов сигналов, которые обнаруживаются примерно в 20% от опухолей.
Факторы транскрипции

Факторы транскрипции — это белки, обнаруженные в ядре клетки, которые в конечном итоге получают сигналы от рецепторов фактора роста.
Программированные регуляторы смерти клеток
Нормальные клетки имеют предопределенный срок жизни, а разные гены регулируют их рост и смерть. Клетки, которые были повреждены или имеют аномальный клеточный цикл, могут развиваться в раковые клетки. Обычно эти клетки разрушаются посредством процесса, называемого запрограммированной клеточной смертью (апоптоз). Клетки, которые развились в раковые клетки, однако, не подвергаются апоптозу. Мутированные протоонкогены могут ингибировать смерть аномальных клеток, что может привести к образованию и распространению рака. Онкоген bcl-2, например, ингибирует гибель клеток в раковых клетках иммунной системы. Механизмы трансформации протоонкогена в онкогены. В большинстве случаев неизвестно, что приводит к тому, что конкретный протоонкоген превращается в онкоген. По-видимому, существуют экологические триггеры, такие как воздействие токсичных химических веществ. Также появляются генетические триггеры, поскольку изменения в других генах в конкретной клетке могут вызывать изменения в протоонкогенах. Однако механизмы, с помощью которых протоонкогены превращаются в онкогены, лучше понятны.
Протоонкогены трансформируются в онкогены через:
- мутацию
- хромосомную транслокацию
- амплификацию гена.
Крошечное изменение, называемое мутацией, в протоонкогене может превратить его в онкоген. Мутация приводит к онкогену, который продуцирует белок с аномальной структурой. Эти мутации часто делают белок устойчивым к регуляции и вызывают неконтролируемую и непрерывную активность белка. Семейство онкогенов RAS, обнаруженное в приблизительно 20% опухолей, является примером онкогенов, вызванных мутациями.
Некоторые протоонкогены участвуют в передаче сигналов между клеточной мембраной и ядром. Например, часть онкогенов является факторами клеточного роста, и при нарушении их регуляции происходит постоянная стимуляция тех или иных процессов. Другие протоонкогены — это рецепторы для факторов тканевого роста. Наиболее полно изучены RAS-онкогены и связанные с ними гены, участвующие в передаче сигналов между клеточной мембраной и ядром. Эти гены активируются при точечной мутации, что приводит к постоянной активации белков-передатчиков сигнала вне зависимости от наличия соответствующего лиганда или рецептора. Часть проонкогенов является факторами транскрипции и, взаимодействуя с ДНК, регулирует транскрипцию генов.
Супрессоры опухолевых генов. При раке толстой и прямой кишки отмечаются большие потери генетического материала. Эти участки выпадения ("горячие" локусы) являются местами сосредоточения супрессивных генов, потеря которых и приводит к развитию рака толстой кишки. Наиболее важный из них — ген р53, в норме предотвращающий начало нового периода синтеза ДНК и деления клетки.
ФУНКЦИИ КЛЕТОЧНЫХ ОНКОГЕНОВ


Из: Hunter T. Cooperation between oncogenes. Cell, 64: 249, 1991
Регуляторный ген (р53), предотвращающий излишнюю пролиферацию клеток, служит супрессивным геном для опухолевых клеток. Характерно, что активация супрессорных генов происходит в два этапа. В ядре клетки существует по две копии любого гена. Супрессивные гены обладают так называемым "доминирующим" действием, когда для супрессии пролиферации клеток достаточно лишь одной полной копии гена. Теория супрессорных генов согласуется с теорией наследственной предрасположенности к возникновению рака. Инактивация одной из двух аллелей может быть фенотипически "немой" (т. е. не происходит развития злокачественного фенотипа), если достаточно одной (второй) аллели для сохранения незлокачественного фенотипа клеток. Но у таких людей особенно высок риск развития рака, т. к. при одной лишь мутации супрессорных генов клетки трансформируются в раковые. В таблице 10-2 приведены наиболее важные гены тканевой супрессии (TSGs).
Образование запретных клонов.
Периодически отдельные клетки мутантного клона подвергаются дополнительной мутации, повышающей их выживаемость и способность к росту. Такие клетки постепенно вытесняют из популяции родительские клетки. Увеличение клона мутированных клеток обычно происходит при появлении новых мутаций, которые увеличивают выживаемость клетки. При наблюдении за развитием опухоли на ранней стадии в клетках выявляется определенное количество мутаций, число которых возрастает с течением времени. На Рис. 10-1 показана прогрессия опухоли на фоне появления клонов с новыми характеристиками.
ПРИМЕРЫ ГЕНОВ, ПОДАВЛЯЮЩИХ РОСТ ОПУХОЛИ У ЧЕЛОВЕКА

Многоступенчатый канцерогенез. Концепция генетической основы развития рака толстой кишки была разработана Vogelstein с соавторами. Согласно этой концепции процесс протекает медленно, и в него вовлечено множество генов.
Количество генетических повреждений опухолей постепенно увеличивается по мере роста. Часть повреждений появляется только на ранних стадиях, часть — только на поздних. Еще до конца не ясна последовательность появления опреде-

Рис. 10-1. Опухолевый рост. На данной схеме представлена типичная последовательность канцерогенеза. На первой стадии показано действие канцерогена. После накопления критической массы мутировапных генов (единой точки зрения на их количество и локализацию нет) клетка превращается в опухолевую. На второй стадии происходит разрастание клона мутировавших клеток с формированием доброкачественной опухоли. Без каких-либо дополнительных воздействий доброкачественная опухоль способна сохранять свою доброкачественность. Но при дополнительном воздействии она трансформируется в злокачественную и приобретает способность к инвазивному росту и метастазированию (стадия 3). Не существует единой точки зрения на природу этого воздействия. Злокачественная опухоль склонна к инвазивному росту с повреждением органов хозяина. Генотип злокачественных клеток очень нестабилен, поэтому их клон оказывается гетерогенным (стадии 4 и 5). Клинически такая гетерогенность приводит к формированию отдаленных метастазов.

Схема последовательных генетических изменений
в многоступенчатом канцерогенезе при раке толстой кишки с дополнениями по Fearon, Vogelstcin. Сверху над схемой показаны мутации, ведущие к потере гетерозиготности (П1), лежащие в основе развития колоректального рака
ленных повреждений в опухолевых клетках, но уже выяснено, что при различных типах рака возможны разные мутации и хромосомные делеции. Для выявления потери участков хромосом используются лабораторные методы, а процесс утраты генетической информации был назван "потерей гетерозиготности".
В ходе своего развития опухолевая клетка кишечника человека претерпевает определенные изменения (рис. 10-2). Гипометилирование ДНК происходит на ранних стадиях возникновения канцерогенеза в аденомах. В клетках небольших аденом могут происходить как мутации, так и выпадения участков хромосом, например изменения на 5q хромосоме при аденоматозном полипозе кишки. Обнаруживаемые в крупных аденоматозных полипах мутации генов K-RASvip53 играют заметную роль в поддержании опухолевого роста. Делеции генов супрессоров опухолевого роста на 18q (встречается при раке толстой кишки) и 17р (локус гена р53) происходят на поздних стадиях и означают малигнизацию процесса. Несмотря на наличие сходства канцерогенеза в толстой кишке и в других отделах желудочно-кишечного тракта, последовательность изменений, а также вовлеченные в процесс гены различны. Детали этих процессов только начинают исследоваться.
Механизмы, замедляющие опухолевую прогрессию. В происхождении опухолей толстой кишки, помимо выпадения супрессорных генов, задействованы и другие механизмы генетических изменений. Микросателлиты — участки ДНК, используемые для картирования генов хромосом. Обычно микросателлиты полностью копируются при репликации клеток, но в некоторых опухолевых клетках отмечается незначительное изменение их длины. Микросателлиты в геноме могут иметь до 103 копий. Было замечено, что в некоторых случаях рака толстой кишки эти микросателлиты копируются не полностью. Наличие "нестабильных" микросателлитов в геноме опухолевых клеток говорит об утрате ими способности адекватно копировать гены. Такое нарушение процессов исправления мутаций считается еще одним механизмом канцерогенеза. Примерная схема этого механизма приведена на Рис. 10-3.
Читайте также: