
Мутации генов при раке толстой кишки
Биологические функции генов семейства RAS
Одно из ключевых свойств любой опухоли — это нарушение баланса между клеточным делением, т.е. размножением клеток, и клеточной гибелью. Для того, чтобы процесс деления осуществлялся в норме, необходимо поступление верного сигнала в клеточное ядро. Подобным сигналом являются специальные белки – факторы роста. Они прикрепляются к определённым рецепторам на поверхности клеточной оболочки и запускают внутри клетки ряд последовательных биохимических реакций. Результатом становится производство и накопление внутри клетки белков, которые необходимы для дальнейшего деления.
Необходимо отметить, что активирующие мутации не только в этих генах, но и в других звеньях сигнальной цепочки могут приводить к постоянной стимуляции клеточного деления. К таким событиям относятся, например, мутации в генах EGFR, BRAF и др. Упомянутые генетические повреждения в разной степени свойственны опухолям различных органов. Например, мутации в генах семейства RAS (KRAS, NRAS, HRAS) встречаются при раке поджелудочной железы, толстой кишки, лёгкого, кожи и т.д. Мутации EGFR характерны для немелкоклеточного рака легкого, а повреждения BRAF с наибольшей частотой обнаруживаются при меланоме.
В каких случаях нужно сделать тест на мутацию в генах KRAS и NRAS?
Тестирование опухоли на наличие мутаций в генах KRAS и NRAS выполняется пациентам с опухолями толстой кишки. Тест позволяет лечащему онкологу решить вопрос о возможности использования в терапии антител к определенному фактору роста и деления клеток ‒ EGFR. Данные препараты – панитумумуаб или цетуксимаб ‒ блокируют расположенные на мембране клеток рецепторы эпидермального фактора роста (EGFR) и препятствуют росту опухоли.
Более того, существуют сведения о том, что ошибочное назначение анти-EGFR терапии пациентам, у которых мутация в перечисленных генах не выявлена, может ускорять рост опухоли. Частота мутаций в гене KRAS при опухолях толстой кишки достигает 50%. Ещё около 10-20% приходится на мутации в генах NRAS и BRAF. Таким образом, при правильном обследовании лечение антителами к EGFR должно назначаться не более 30-40% пациентов с раком толстой кишки, в остальных случаях используются другие схемы терапии.
Что делать, если в опухоли толстой кишки обнаружена мутация в гене KRAS или NRAS?
Обнаружение мутации в гене KRAS или NRAS является абсолютным противопоказанием к использованию цетуксимаба или панитумумаба, т.к. присутствие этих мутаций полностью препятствует противоопухолевому действию данных препаратов. Как упоминалось выше, при ошибочном назначении антител к EGFR пациентам с мутациями в генах RAS может наблюдаться ускорение роста опухоли – именно поэтому полноценное исследование данных генов является обязательным условием для подбора правильной терапии. В случае наличия мутаций в генах KRAS и NRAS в клетках опухоли успешно используются другие разновидности лечения.
Как сдать анализ на мутации в гене KRAS, NRAS, EGFR, BRAF?
Чтобы провести молекулярно-генетическое тестирование, специалистам необходимы опухолевые клетки. Они могут быть получены либо при биопсии, либо в ходе хирургической операции по удалению новообразования. При этом, материал для исследования должен быть подготовлен определенным образом. В противном случае тестирование будет невозможно.
В ходе первичного обследования онкологическому пациенту практически всегда выполняют биопсию, на основании которой происходит патоморфологическое подтверждение диагноза. Для этого полученные клетки пациента проходят многоэтапную химическую обработку. В результате из них создаётся специальный парафиновый блок. С одной стороны, он необходим для получения качественного тонкого среза (толщиной 5 мкм) с целью патоморфологической диагностики. С другой стороны, в правильно подготовленном парафиновом блоке молекулы ДНК надёжно сохраняются на протяжении десятилетий.
Аналогичные манипуляции патологи проводят в отношении опухолевых тканей, удалённых во время операции. Правильное выполнение процедуры фиксации тканей позволяет использовать образцы опухолей для молекулярно-генетического исследования ДНК спустя месяцы и годы после заливки образца в парафин.
Идеальным набором для молекулярно-генетического исследования является следующий комплект: парафиновый блок c тканью опухоли и одно стекло, окрашенное специальными красителями (гематоксилином и эозином). Всё перечисленное хранится в патологоанатомических архивах медицинских учреждений, а окраска гематоксилином и эозином – основная окраска, используемая в современной патоморфологической диагностике. Если медицинское учреждение по какой-либо причине не может предоставить блоки, то для молекулярно-генетического тестирования достаточно 5-10 неокрашенных срезов ткани опухоли на непокрытых стёклах толщиной 3-5 мкм и одно стекло, окрашенное гематоксилином и эозином.
Требования к упаковке материала перед транспортировкой
- Закрывающийся пластиковый пакет или контейнер, либо картонная коробка
- Полное соответствие номеров отправляемых блоков и стёкол в направлении на тест и копии патоморфологического заключения.
- Лабораторные стекла должны быть обёрнуты плотной бумагой для избежания повреждений.
- Хранение производится при комнатной температуре, не допустим нагрев блоков и стёкол выше +50 о С.
В настоящее время многие молекулярно-диагностические исследования выполняются за счет средств территориальных фондов ОМС регионов России (как Санкт-Петербурга, так и остальных субъектов РФ).
![]()

У четверти людей с онкологическими заболеваниями кишечника обнаруживается отягощенный семейный анамнез. Примерно 10 % эпизодов рака толстой кишки связаны с наследственным фактором. Риск развития опухоли высок при наличии у пациента аденоматозного полипоза и синдрома Линча.
Причины наследственного рака толстой кишки
Перерождение нормального эпителия в раковые образования связано с определенными мутациями в генах, которые могут быть врожденными (в гене АРС) и приобретенными (амплификация гена MYC, точечные мутации гена KRAS, делеции определенных участков хромосом 5, 8, 17, 18).
В 50 % случаев наследственного рака и крупных аденоматозных образований толстой кишки выявляют точечные мутации гена KRAS. В 75 % случаев аденокарционом обнаруживается делеция 17р (в гене TP53), при 30 % крупных аденоматозных полипов и аденокарцином — делеция длинного плеча 5q (в гене APC).
Наследственный аденоматозный полипоз, связанный с мутацией гена-супрессора опухолевого роста APC (в локусе 5q21), грозит развитием рака толстой кишки к 55 годам. Синдром Линча, обусловленный мутациями в генах MSH2 , MLH1 и MSH6 (хромосомы 2 и 3) или гена PMS2 (хромосома 7), характеризуется ранним развитием новообразований (20 лет) и менее агрессивным течением болезни.
Диагностирование наследственного рака толстой кишки
Симптомы заболевания включают в себя: кровь в кале, признаки анемии, кишечной непроходимости, болевой синдром. Больной теряет вес, ухудшается аппетит, появляется слабость, повышается температура тела.
Опухоль обнаруживается во время колоноскопии с биопсией. О присутствии опухолевого процесса могут рассказать исследование кала на скрытую кровь, анализы крови на онкомаркеры, а также гастроскопия, УЗИ, рентгенография, сцинтиграфия.
- возраст пациента на момент обнаружения новообразования — не старше 50 лет;
- множественные раковые образования в кишечнике или других органах, которые характерны для наследственного неполипозного рака толстой кишки;
- наличие у больного признаков генетической нестабильности — Microsatellite Instability (MSI);
- в семье пациента имеется минимум 2 случая опухолей кишечника родственников младше 50 лет;
- минимум 3 случая онкообразований кишечника в семье, вне зависимости от возраста обнаружения патологии.
Опухоль, удаленная во время операции, подлежит обследованию на предмет:
- генетической нестабильности MSI;
- иммуногистохимического анализа белков Mismatch Repair (MR).
Если результаты одного из этих анализов указывают на наследственный рак толстой кишки, назначают генетическое тестирование (анализ крови на мутации в генах DNA Mismatch Repair Genes). Если тест подтверждает присутствие патологии, то такое исследование рекомендуется и родственникам больного.
Лечение наследственного рака толстой кишки
Проктоколэктомия — предпочтительный метод терапии заболевания. При поражении лимфоузлов показана лимфаденэктомия, в случае отдаленных метастазов — паллиативные вмешательства.
Профилактика
Носители мутации находятся в группе риска по наследственному раку толстой кишки и нуждаются в регулярных скрининговых обследованиях для раннего выявления онкопатологии (колоноскопия, анализы на онкомаркеры СА125, СА19-9, карциноэмбриональный антиген и др.).
Рак толстой кишки занимает 2-е место по смертности от злокачественных новообразований. По мере старения общества показатель летальности от рака этой локализации будет увеличиваться. Это неудивительно, если вспомнить об этиологии заболевания и особенностях патоморфологических изменений толстой кишки: ее слизистая оболочка представлена одноклеточным эпителием с быстрым периодом полного обновления и постоянной экспозицией к воздействию канцерогенных факторов.
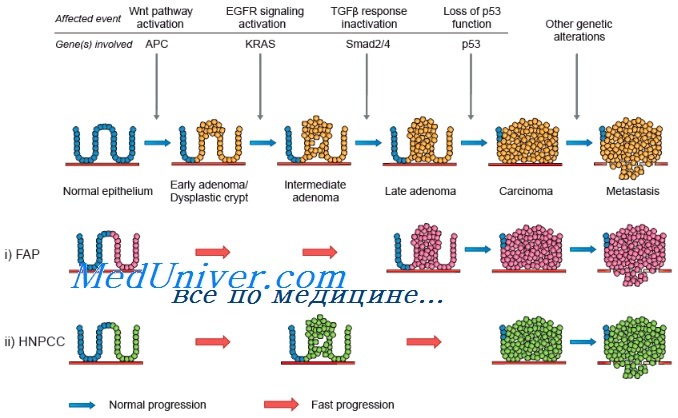
Следовательно, ДНК этих клеток постоянно и быстро делится и реплицируется. В результате вероятность накопления ошибок и злокачественной трансформации со временем увеличивается. Модель рака толстой кишки, разработанная Vogelstein и соавт., иллюстрирует эти механизмы. Мутации гена-супрессора аденоматозного полипоза толстой кишки (adenomatous polyposis coli, АРС) ведут к разрастанию полипов толстой кишки в раннем возрасте. Для уточнения характера любого заболевания толстой кишки можно легко получить образец ткани путем биопсии. По мере роста полипа он становится доступен для обследования гастроэнтерологом с использованием ряда исследований. Рак толстой кишки хорошо изучен также потому, что существуют семьи, члены которых подвержены редкому заболеванию — семейному аденоматозному полипозу толстой кишки. У этих больных толстая кишка покрывается сотнями и тысячами полипов, один или несколько из которых, как правило, малигнизируются в зрелом возрасте.
Именно у этой категории больных впервые был обнаружен ген АРС. В большинстве спорадических опухолей толстой кишки выявляют соматические мутации гена АРС. В более редких семейных случаях отмечается мутация гена АРС, передающаяся по наследству и в связи с этим присутствующая во всех эпителиальных клетках кишечника, что и предрасполагает к развитию полипов. Соматическая мутация ведет к активации онкогена ras и ускорению роста полипа, который, тем не менее, еще остается доброкачественным. Однако с годами полип может приобрести мутации генов-супрессоров DCC и ТР53, которые ведут к автономному неконтролируемому росту злокачественных клеток рака толстой кишки.
Семейная предрасположенность к раку толстой кишки достаточно широко распространена и делится на две группы: с образованием полипов и без них. Полипозы включают семейный аденоматозный полипоз толстой кишки, синдром Пейтца— Егерса, семейный ювенильный полипоз и гиперпластический полипоз. Риск рака толстой кишки у всех больных с названными синдромами повышен. Например, медиана возраста появления рака толстой кишки при семейном аденоматозном полипозе составляет 40 лет.
К синдромам без полипоза относится наследственный неполипозный колоректальный рак (ННПКРР). У этих пациентов злокачественную опухоль толстой или прямой кишки выявляют в возрасте около 40 лет. У большинства больных ННПКРР идентифицируют генетические дефекты в системе репарации ДНК. Мутации происходят в основном в гене MLH1, однако результаты последних исследований свидетельствуют о частом поражении таких генов, как MSH2 и MSH6. ННПКРР сопровождается и другими онкологическими заболеваниями: раком эндометрия (РЭ), раком яичника (РЯ), раком молочной железы (РМЖ) и опухолями нервной системы.
Семейный рак толстой кишки составляет примерно 3—5 % всех случаев рака этой локализации. Однако риск рака толстой кишки также повышен у родственников больных любой формой этого заболевания. Поэтому в большинстве случаев рака толстой кишки можно наблюдать, по крайней мере отчасти, наследственный компонент. Факторы окружающей среды играют важную роль в развитии заболевания; полученные данные указывают, что диета с высоким содержанием клетчатки может снижать вероятность развития рака толстой кишки. Фактор риска — рацион питания с высоким содержанием жира и красного мяса.
*Локализация на хромосоме
Молекулярные изменения, происходящие при раке толстой кишки, хорошо исследованы Vogelstein и соавт., подробно описавшими прогрессию нормального эпителия в рак. Первыми генами, для которых была установлена связь с раком толстой кишки, стали гены семейства ras, изначально выявленные в вирусах саркомы крыс. Эти гены наиболее часто обнаруживают в промежуточных полипах толстой кишки без дисплазии. В аденомах на ранних стадиях мутации ras встречаются редко, но в промежуточных полипах — регулярно, что свидетельствует о важной роли этих генов в раннем развитии рака толстой кишки.
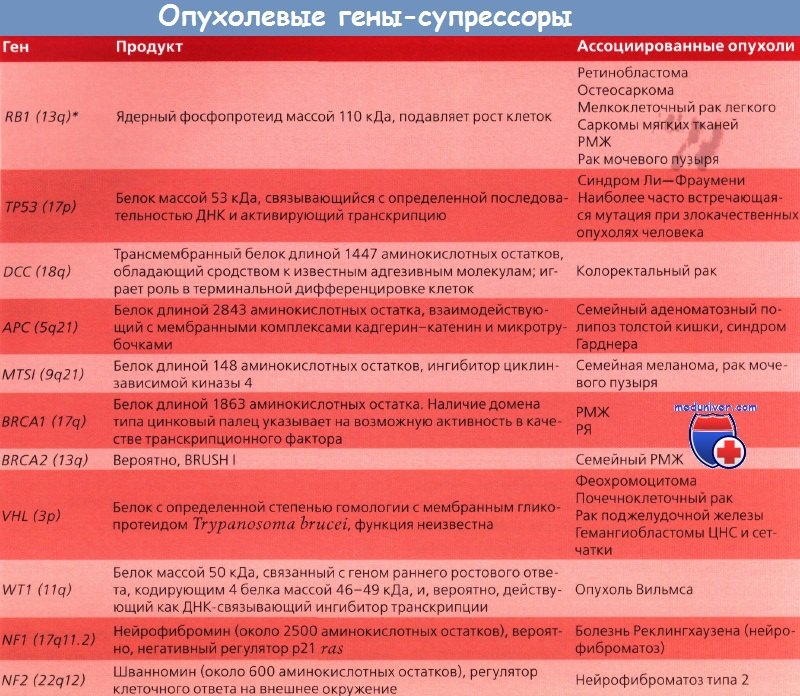
Опухолевый ген-супрессор АРС принимает участие на самых первых этапах канцерогенеза при раке толстой кишки и был выявлен в исследованиях по изучению утраты гетерозиготности, показавших часто встречающуюся утрату длинного плеча хромосомы 5 (5q). Еще один ген, связанный с развитием рака толстой кишки, — ген-супрессор ТР53, расположенный на коротком плече хромосомы 17 (17р). Мутация этого гена — относительно позднее событие при раке толстой кишки. ТР53 — важный ген в контроле клеточного цикла и апоптоза. Утрата гена-супрессора DCC на длинном плече хромосомы 18 (18q) происходит на промежуточных и поздних этапах канцерогенеза при раке толстой кишки. Этот ген кодирует белок, ответственный за клеточную адгезию.
Ген DPC4, также расположенный на длинном плече хромосомы 18, вовлечен в сигнальную систему, связанную с трансформирующим фактором роста b.
При развитии рака толстой кишки происходит последовательное приобретение ряда мутаций. Для развития злокачественной опухоли обязательного прохождения всех этапов не требуется, однако предполагается, что необходимо по меньшей мере 6 или 7 генетических событий. У больных наследственным неполипозным колоректальным раком (ННПКРР) эти мутации развиваются вследствие неспособности эпителиальных клеток слизистой оболочки кишечника выявлять и устранять ошибки, происходящие во время нормального клеточного деления. Исследования Vogelstein и соавт. продемонстрировали модель, которая может быть применена и к другим опухолям.
Генетика семейного рака кишечника
Рак часто наблюдается у членов одной семьи, объективно подтверждена наследственная природа некоторых злокачественных опухолей. Есть точка зрения, что наследственная предрасположенность — самая вероятная причина всех онкологических заболеваний, и только дело времени, чтобы наука точно установила, мутация какого гена за какой конкретный рак отвечает. Но уже сейчас наследственную передачу рака можно прервать.
Если у человека возникло онкологическое заболевание, очень важно выяснить, есть ли в его роду другие случаи злокачественных новообразований. Семьям, в которых имеется более одного такого случая, нужно пройти консультацию врача-генетика, чтобы понять, есть ли в семейной истории основания для подозрений на наследственный характер патологии. Особенно настораживающим признаком будет онкологическое заболевание в нескольких поколениях семьи. Одним из основных методов работы врача-генетика является составление родословных. Другая важная часть медико-генетической консультации — осмотр и опрос пациента: наследственные заболевания нередко проявляются специфическими признаками.
Принципиальным отличием наследственного рака является возможность его прогнозировать путем выявления патогенных мутаций. На первом этапе семьям, в которых имеется более одного случая развития рака, рекомендуется пройти консультацию врача-генетика, по результатам которого можно будет понять, есть ли в семейной истории основания для подозрения на наследственный характер патологии.
Если в процессе консультации возникают подозрения на наследственную природу заболевания, то следующий этап — целенаправленное генетическое тестирование, поиск мутаций, которые могут вызывать конкретное заболевание. Одни исследования позволяют обнаружить изменения в самом гене, другие — в белке, который кодируется измененным геном. Один ген может претерпеть до 300 мутаций.
В последние годы найдены мутации, ответственные за возникновение и развитие рака молочной железы, яичников, толстой кишки и др. Цель генетического тестирования, или скрининга,— выявить риск возникновения заболевания до появления симптомов. Это дает возможность в одних случаях провести своевременное лечение, в других — рекомендовать меры, позволяющие избежать передачи наследственного заболевания потомству. Мутации генов найдены для нескольких видов рака, тесты на некоторые из них уже используют в клинике — например, тесты на рак груди и кишечника.
От предков или не от предков
Все онкологические заболевания имеют генетическую природу, поскольку при раке гены, отвечающие за правильное деление клетки, повреждены. Но в одних случаях имеют место наследственные мутации, а в других — приобретенные. Результатом повреждения (мутации) гена во всех случаях является бесконтрольное неограниченное деление клеток, что и является сутью ракового процесса.
Несмотря на то что онкологические заболевания имеют генетическую природу, только 10–15% из них передаются по наследству. Почему важно знать, наследственный или ненаследственный рак? Потому что если установлена его наследственная природа, то есть выявлена мутация, вызвавшая его, то известен прогноз и понятна тактика в отношении самого больного и его родственников. Особенно отчетливо наследование мутации прослеживается в случаях так называемого семейного рака молочной железы и яичников, при семейном аденоматозном полипозе и различных опухолевых синдромах (Линча — рак толстой кишки, Ли-Фраумени — разнообразные саркомы и др.). Многие люди, сами будучи здоровыми, являются носителями мутаций, приводящих к наследственным заболеваниям. Если носители одной и той же мутации — оба родителя, заболевание становится неизбежным. Генетическое тестирование позволяет это выявить.
Следует подчеркнуть, что наличие мутации не означает заболевания. Мутация может сидеть в гене много лет до того, как начнет развиваться опухоль. Но, зная про мутацию, врачи могут назначить рациональный режим обследования и профилактического лечения.
Например, у женщин—носительниц гена BRCA1 в 95% случаев в течение жизни разовьется рак груди и в 65% — рак яичников, причем часто рак развивается в молодом возрасте, до 50 лет. Это означает, что носительница должна все время находиться под наблюдением, а в некоторых случаях целесообразно ставить вопрос о профилактическом удалении груди и (или) яичников. У всех на слуху история Анджелины Джоли, которая настояла на удалении обеих молочных желез, поскольку у нее обнаружили мутацию гена BRCA1.
Специалисты знают результаты исследования ткани удаленных молочных желез у 54 шведских женщин—носительниц этого гена в возрасте до 51 года. Ни у одной из них обследование не показывало опухоли груди до операции, но гистологическое изучение удаленной ткани выявило наличие раковых клеток у пяти (10%!) из них.
К профилактической хирургии прибегают и при семейном аденоматозном полипозе, при котором вероятность развития рака толстой кишки после 40 лет достигает 100%, и при других онкологических заболеваниях, если установлена онкогенная мутация.
Понятно, что женщины с отрицательным результатом теста на мутации генов BRCA1 и BRCA2 не застрахованы от спорадического рака груди и яичников. Однако вероятность его возникновения несопоставимо ниже, чем у женщин с положительным тестом.
Женщине следует заподозрить у себя предрасположенность к наследственному раку груди, пройти консультацию врача и генетика и генетическое тестирование, если в семье:
— было более одного случая рака груди или яичников по женской линии (у матери, бабушки, тетки, сестер и т. д.);
— заболевание было диагностировано в молодом возрасте (до наступления климакса);
— были случаи рака груди у мужчины;
— были больные c множественными опухолями (например, у одного человека — рак груди, толстой кишки, матки, рак поджелудочной железы и т. д.);
— были случаи двустороннего рака обеих молочных желез или обоих яичников.
Тестирование и его последствия
Генетическое тестирование имеет несколько преимуществ. Отрицательный результат может принести человеку облегчение, избавить от страха ожидания тяжелой болезни, от которой, возможно, погибли его близкие, а также от регулярных обследований, которые должны быть обязательны в семьях с высоким онкологическим риском. Положительный результат дает человеку возможность принимать обдуманные решения о будущем своем и своего потомства.
Сегодня возможна профилактика наследственного рака, то есть возможность не передать от родителей потомству ген, несущий опасную мутацию. Метод, который позволяет это сделать, называется преимплантационная генетическая диагностика (ПГД). Он заключается в следующем: для пары выполняют ЭКО, проводят генетическую диагностику полученных эмбрионов и переносят в матку женщины только те из них, в которых нет онкогенных мутаций. У родившегося ребенка их не будет, а значит, не будет и наследственного рака.
Открытое письмо Анджелины Джоли, New York Times, 14 мая 2013 года
ПГД проводится не на всем эмбрионе, а на нескольких клетках, которые получают путем его биопсии. Доказано, что биопсия не оказывает влияния на здоровье и состояние ребенка. Другими словами, ПГД не снижает частоту наступления беременности и безопасна для будущего ребенка.
Кроме мутаций, отвечающих за развитие рака груди и яичников, установлены мутации, несущие предрасположенность к меланоме, раку желудка, матки, предстательной, поджелудочной и щитовидной железы, толстой и прямой кишки. Если мутация определена и в семье есть люди, которые хотят иметь ребенка, важно, чтобы они знали о возможности предотвратить передачу следующим поколениям этой мутации и связанного с ней рака с помощью ЭКО и ПГД.
Об этом мы поговорили с онкогинекологом, хирургом Владимиром Носовым, руководителем Клиники гинекологии и онкогинекологии Eвропейского медицинского центра – первой клиники в России, где персонализированная терапия онкогинекологических заболеваний стала стандартной практикой.
В своём нормальном состоянии эти гены участвуют в восстановлении ДНК после различных повреждений, тем самым защищая клетки от опухолевого перерождения. Если возникает мутация в этих генах, здоровые клетки оказываются не защищенными и сами могут становиться злокачественными. Вероятность заболеть раком груди при носительстве мутации гена BRCA 1/2 колоссальная — до 80%(в общей популяции у женщин без мутации — около 10-12%), риск заболеть раком яичников — до 40-45 %( в популяции —около 1,5%) .
В большинстве случаев назначение этих препаратов после первой линии химиотерапии обеспечивает ремиссию около 3 лет – это огромное достижение, еще никогда в онкогинекологии ремиссия при 3-4 стадии заболевания не продлевалась каким-либо лекарством на столь длительный срок.
Дальнейшие исследования позволили выяснить, что мутации могут быть не только герминогенными, то есть присутствующими во всех клетках организма. Дополнительные 15-20% мутаций генов BRCA происходят только в клетках опухоли, но в крови и других клетках организма их нет. Эти мутации называют соматическими. Они не передаются по наследству, не увеличивают риск развития других онкологических заболеваний, но пациенты, у которых обнаружены мутации в клетках опухоли, также являются кандидатами для лечения ингибиторами PARP.
В Институте онкологии EMC мы предлагаем всем пациентам с раком яичников провести полное секвенирование генов BRCA опухоли и крови. Это позволяет подобрать наиболее эффективную персонализированную терапию. Если речь идет о наследственной мутации – мы рекомендуем в обязательном порядке генетическое обследование детям, сестрам, братьям, родителям, а самим пациенткам-носителям мутации – также пройти дополнительный скрининг на рак молочных желез, риски которого колоссально повышены.
Плохое наследство
Наследственная мутация передается детям с вероятностью 50%, причем как по женской, так и по мужской линии. Носителям мы рекомендуем специальную программу наблюдения и профилактические мероприятия для снижения риска онкологических заболеваний, а также обсуждаем с ними вопросы сохранения репродуктивной функции.
Например, на днях я оперировал пациентку 57 лет с раком яичника. На плановой гистологии был подтвержден злокачественный характер опухоли. Мы провели генетическое исследование опухоли, выявили мутацию BRCA1. Затем было выполнено полное генетическое исследование по крови, чтобы понять, является ли мутация соматической (присутствующей только в опухоли) или герминогенной (наследственной). Выяснилось, что мутация наследственная. Мы рекомендовали пройти обследование двум дочерям пациентки, которые, к сожалению, унаследовали эту мутацию. Женщины-близнецы, им сейчас 31 год, обе еще не планировали беременность и роды. Я рекомендовал им обратиться к репродуктологу, провести стимуляцию и заморозить яйцеклетки, а в 35 лет, именно с этого возраста риски рака яичников начинают расти, удалить профилактически яичники и маточные трубы. В этом случае мы сохраняем матку, и в будущем они смогут выносить своих биологических детей.
Более того, во время ЭКО можно провести предимплантационную диагностику и подсадить эмбрионы, не унаследовавшие мутацию. Таким образом, будущее поколение уже будет защищено.
Рак эндометрия (рак тела матки) – самое распространенное онкогинекологическое заболевание у женщин. Сегодня подходы к его лечению также меняются благодаря персонализированной терапии.
До недавних пор считалось, что существует два типа рака эндометрия. Наиболее частый, первого типа, обычно возникает у полных пациентов, часто с сопутствующими диабетом и гипертонией. Второй – серозный, более агрессивный, не связанный с избытком эстрогенов. На основании клинической картины врачи принимали решение о необходимости дополнительного лечения после операции. Сегодня, благодаря лучшему пониманию биологии опухоли, мы знаем, что этих типов не два, а четыре. И для каждого из них предусмотрено определенное лечение. Чтобы определить, с каким типом рака эндометрия мы имеем дело, достаточно для начала провести иммуногистохимическое исследование.
Каждую опухоль эндометрия вне зависимости от стадии, мы тестируем на наличие определенных молекул, указывающих на благоприятный или менее благоприятный прогноз заболевания. Например, наличие мутации гена P53 говорит о менее благоприятном прогнозе. В этом случае мы рекомендуем не только наблюдение, но и дополнительное лечение с помощью химио-или лучевой терапии.
Некоторые раки матки, так же, как и некоторые раки яичников и молочной железы, имеют в своей основе генетический синдром – синдром Линча. Если мы находим проявления синдрома Линча в опухоли, мы направляем пациентов на полноценное генетическое тестирование. Это важно, потому что рак матки – не единственное заболевание, к которому предрасположены носители мутаций, вызывающих синдром Линча. В частности, у них повышен риск рака толстой кишки в молодом возрасте.
Часто первым возникает рак матки, через какое-то время развивается рак толстой кишки.
Поэтому носителям синдрома Линча рекомендуют начинать скрининг на рак кишки не в 45-50, а гораздо раньше — с 30 лет и делать колоноскопию раз в 6 или 12 месяцев, чтобы не пропустить развитие заболевания.
Выявление синдром Линча у пациентки с раком матки может повлиять и на лечение.
При поздних стадиях пациентам с синдромом Линча мы назначаем специфическую иммунотерапию препаратом пемпролизумаб, что позволяет улучшить прогнозы пациентов.
Генетическое профилирование опухоли – это колоссальный прорыв, который позволил нам подойти к полностью персонализированной терапии в онкологии, основанной не только на диагнозе, но и на понимании биологии опухоли. Для пациентов — это возможность получить точное узкоспециализированное лечение, дающее лучшие результаты, а в случае наследственных раков — возможность защитить будущие поколения от опасных заболеваний.
К самым большим достижениям фундаментальной науки в рассматриваемой области следует отнести идентификацию генетических изменений при спорадическом (ненаследственном) раке толстой кишки, которая дала возможность приступить к разработке генной терапии этого заболевания во многих онкологических центрах и научно-исследовательских лабораториях крупнейших фармацевтических компаний.
Впервые модель колоректального канцерогенеза была представлена в литературе Vogelstein et.al в 1988 году. В последующем эта модель подверглась уточнениям и сейчас признается, что она правильно аккумулирует генетические изменения и спорно описывает последовательность этих изменений. Модель колоректального канцерогенеза и мишени генной терапии представлены ниже.
РТК развивается от 8 до 15 лет. Это многоступенчатый процесс, характеризующийся на разных этапах специфическими морфологическими и молекулярными изменениями.
Гиперпролиферация нормального эпителия является началом РТК (образование фокусов диспластических аберрантных крипт).
Мутации супрессорного гена АРС ответственны за этот этап канцерогенеза. Они были найдены на ранних стадиях РТК в 70%. Японские исследователи продемонстрировали на мышах, что мутация АРС быстро приводит к образованию многочисленных аденом кишечника [Shibata, 1997].
Цитогенетическим выражением мутаций этого гена является потеря одного или двух аллелей в 5 хромосоме. Мутации отмечаются в районе 15-го экзона, в котором осуществляется взаимодействие с геном β-катенин, о чем ниже. Установлено, что главным следствием мутации АРС является гиперактивация Wingless Wnt пути сигнальной трансдукции, в результате чего транскрипируются гены, благоприятствующие возникновению опухолевого роста. Мутации АРС приводят к накоплению β-катенина. Этот ген связывается с транскрипционными факторами TCF/LEF, что приводит к активации циклина D1 промотора клеточной пролиферации. Активируются и другие гены C-Jun, Fra-1, WISP, соединительнотканный фактор роста Суr61 и др., все они являются промоторами клеточного деления.
Образование ранних аденом – второй этап в развитии РТК, его связывают с мутированным колоректальным раковым геном (ММС) и гиперметилированием ДНК. Ген ММС расположен в 5 хромосоме, он играет важную роль в передаче сигнальной трансдукции, его значение при РТК пока лишь является предположением.
Метилирование ДНК необходимо для регуляции экспрессии генов и важно для метаболизма цитозин нуклеотидов. В ДНК клеток аденом содержится меньше метальных групп, чем в клетках нормальной слизистой. Гиперметилирование по современным представлениям дополняет клеточную генетическую нестабильность.
Цитогенетическим последствием гиперметилирования является потеря двух аллелей и инактивация гена hMLH.
Третий этап развития РТК – поздние аденомы – связан с мутацией генов ras. В середине 60-х годов гены этого семейства были впервые охарактеризованы как трансформирующие в канцерогенезе толстой кишки.
Особое значение имеет K-ras, меньшее N-ras. Оба гена являются маркерами опухолевой прогрессии, K-ras расположен в коротком плече 12-й хромосомы, N-ras – в 1-й хромосоме. Vogelstein (1988) обнаружил K-ras мутации у 13% больных с ранними тубулярными аденомами, в 42% у больных с крупными аденомами и в 57% у больных с аденомами с участками инвазивного рака.
Именно Р53 в норме регулирует транскрипцию генов, отвечающих за продолжение клеточного цикла, апоптоз и ингибицию ангиогенеза. Среди этих генов р21, Вах, GADD 45, FAS-Apo, циклин G, TSPI и другие.
Р53 мутирован более чем у 50% больных РТК, мутации его располагаются в экзонах 5-8 и сопровождаются потерей аллеля в коротком плече 17 хромосомы.
Замещение или подавление мутированных генов на разных этапах колоректального генеза – таковы вполне достижимые цели будущего лечения РТК. Экспериментальные разработки ведутся весьма энергично.
Как мы упомянули, поэтапная модель Vogelstein подвергается ревизии и дополнению. В 21 веке внесены существенные изменения в представлении о колоректальном канцерогенезе и ответственности различных мутированных генов. В суммарном виде генетические изменения при РТК характеризуются:
- количественными нарушениями – в клетках РТК меньше ДНК, что обусловлено потерями аллелей в 17р, 18q, 5р, 8р и 22q;
- качественными нарушениями – супрессорные гены Р53, АРС, CMAD4, р16 инактивируются в связи с мутациями, гиперметилированием или потерей аллелей;
- активируются протоонкогены в онкогены (K-ras, BRAF, Р1КЗСА);
- нарушены гены, исправляющие дефекты ДНК, – MLH1, MSH2, MSH3, MSH6;
- активируются тирозинкиназные гены (ТК-гены) NTRK3, FES, KDR, ERHA3, NTPK2, MLK4;
- угнетаются фосфатазные гены: PTPRG, PTPRF, PTPRT, PTPN3, PTPN13, PTPN17.
К 2005 году идентифицированы по генетическим изменениям 2 типа колоректальных опухолей. 1-й тип характеризуется потерей гетерозиготности (LOH). Под этим термином обозначается делеция одного аллеля в опухолевой клетке, преимущественно в длинном плече 18-й хромосомы или коротком плече 17 хромосомы, а также реже в 5q, 8р и 22q хромосомах. Более чем 2/3 РТК принадлежат к этому типу. Почти всегда мутированы гены Р53 и АРС, отмечается частая мутация K-ras и PIK3CA генов. Рак локализуется обычно в дистальной части толстой кишки, опухоли гиперплоидные. Хромосомная нестабильность – характерная особенность этого типа РТК.
2-й тип встречается у 1/3 больных, чаще в проксимальных отделах толстой кишки, его называют микросателлитным локус позитивным раком (MSI-positive). Он характеризуется генной нестабильностью. Не бывает потери аллелей в 17р 18q 5q, редки мутации генов Р53 и АРС, регистрируются мутации рецепторов II типа TGFB, ВАХ, TGF4, каспазы 5, часто мутированы гены BRAF, PIK3CA, характерными являются изменения генов hMSH2, hMLH1, hMSH3, исправляющих дефекты ДНК [Laurent Puig, 2005].
Читайте также: