
Лимфопролиферативный синдром при лейкозах
Аутоиммунный лимфопролиферативный синдром – передающееся по наследству патологическое состояние. Принадлежит к категории гетерогенных. Есть два механизма наследования: аутосомный доминантный и рецессивный. В редких случаях причина – соматические мутации. Лимфопролиферативный синдром может быть приобретенным.
История и факты
Впервые первоначальный х-сцепленный лимфопролиферативный синдром мальчиков был официально признан и оформлен в науке в 1967 году. С 1976 его причисляют к первичному иммунодефициту. Внимание ученых к патологическому состоянию приковано с последних десятилетий прошлого столетия. Уже тогда было выявлено, что базой развития заболевания становится неправильный лимфоцитный апоптоз.

Выявляя особенности аутоиммунного лимфопролиферативного синдрома, ученые установили, что всем больным свойственна неправильная экспрессия рецепторов мембран fasl, CD95. Именно этот нюанс определяет генетически объясняющуюся способность клеток умирать. Патологическое состояние развивается в случае генной мутации, влияющей на апоптоз.
Биология и анатомия: как все происходит?
В медицинской пропедевтике лимфопролиферативный синдром принято делить на несколько разновидностей. Для классификации учитывают особенности генетических отличий конкретного случая. Генные мутации могут затрагивать восьмую и десятую каспазы, CD95, CD178. В то же время стоит отметить, что не существует общепризнанной официальной классификации случаев на группы.
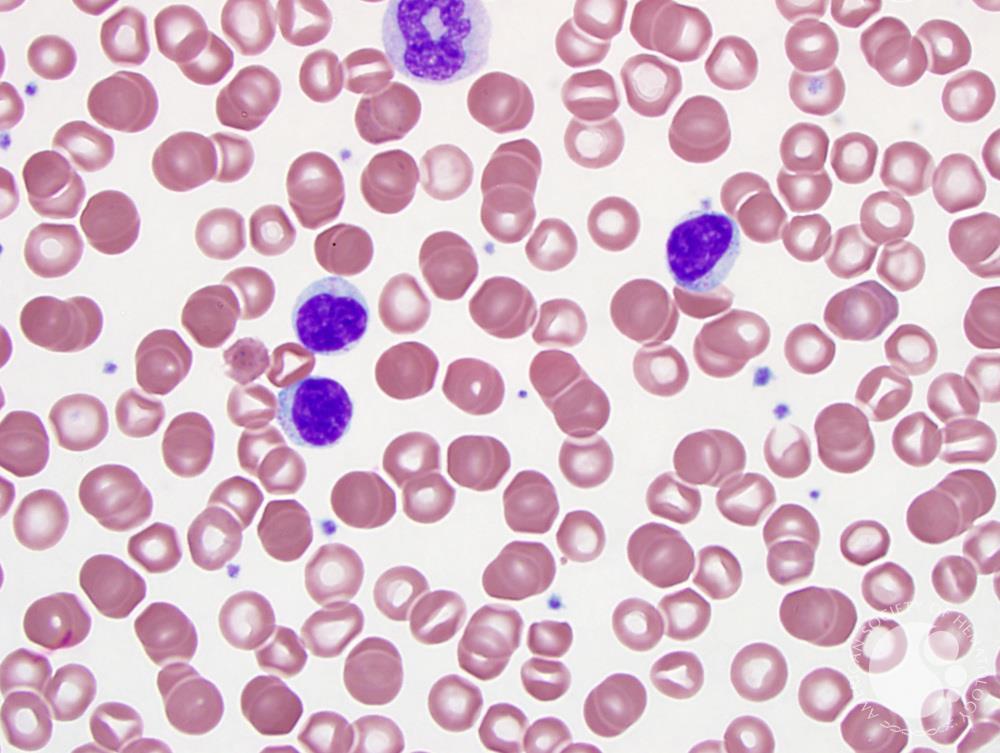
Особенности проявления
Симптомы заболевания исключительно разнообразны. Обычно х-сцепленный лимфопролиферативный синдром выявляют на первых годах жизни, несколько реже – в более старшем возрасте (до пятнадцатилетнего). Ключевой симптом – пролиферация лимфоидной ткани, провоцирующая спленомегалию, лимфаденопатию. Явлениям присущ хронический характер течения. Одновременно больной страдает от проявлений аутоиммунного дисбаланса. Анализы помогают выявить аутоиммунную цитопению. Она возможна в форме нейтро-, тромбоцитопении, анемии. Несколько реже цитопения появляется ранее пролиферации лимфоидных тканей.
Х-сцепленный лимфопролиферативный синдром провоцирует нарушения в работе кроветворной, кровеносной систем. Как правило, фиксируется гепатит аутоиммунной природы. Многие страдают от экземы, гломерулонефрита. Пациентам свойственны увеит, тиреоидит. Приблизительно у каждого десятого со временем формируется лимфома из клеток типа В.
Клинические проявления
Лимфопролиферативный синдром у детей имеет ряд типовых признаков. Наиболее яркий – лимфопролиферация. Процессу присущ доброкачественный характер, патологическое состояние хроническое. Обычно формируется уже в раннем детстве, иногда устанавливается у годовалых малышей. Состояние сохраняется от полугода и более. Вместе с тем наблюдается персистирующее разрастание лимфоузлов периферической лимфосистемы. Для постановки диагноза необходимо выявить такие процессы в трех группах узлов или большем количестве. Узлы плотные, с расположенными поблизости тканями не спаяны. У многих анализы помогают выявить гепатоспленомегалию.
Х-сцепленный лимфопролиферативный синдром у мальчиков проявляет себя аутоиммунными признаками. Классический вариант – анемия, нейтро-, тромбоцитопения. Возможен васкулит. Нередки случаи артрита, гепатита. Больные склонны к увеиту, гломерулонефриту, тиреоидиту. Возможны некоторые другие болезни аутоиммунной природы.
Обратить внимание!
Лимфопролиферативный синдром сопряжен с высокой вероятностью развития злокачественного формирования. Область локализации процесса непредсказуема. Неправильно протекающий апоптоз, работа которого сопряжена с активностью рецепторов Fas, приводит к понижению контроля за процессами разрастания тканей. Растет способность выживать у клеток, переживших патологическую трансформацию. В норме указанный ген – это угнетающий развитие компонентов опухолей фактор.
Чаще заболевание сопровождается формированием лимфом типа В, Т. Кроме того, высока вероятность раковых процессов в молочной железе, кишечном тракте, органах дыхания. Миело-лимфопролиферативный синдром с высокой степенью вероятности может спровоцировать лимфогранулематоз.
При аутоиммунном заболевании пациент склонен к крапивнице, васкулиту. У некоторых отмечается замедленное развитие организма.
Уточнение диагноза
Лимфопролиферативный синдром диагностируют, если установлена не носящая злокачественный характер лимфаденопатия. Возможна спленомегалия. Диагноз ставят при комбинации этих двух явлений или присутствии любого из них, если длительность развития состояния – полгода и больше. При подозрении на диагноз необходимо направить пациента на анализы. В лабораторных условиях устанавливают сбой опосредованного лимфоцитного апоптоза, уточняют концентрацию клеточных структур CD4, CD8 Т: при содержании более 1% можно говорить о патологическом состоянии.
Особенности случая
Далее рассматривается форма заболевания, не связанная с нарушением генетики в период развития эмбриона. Нижеописанное касается приобретенной формы заболевания.
Лимфопролиферативный синдром – симптомокомплекс, который может сопровождать не только лимфолейкоз, протекающий по типовому сценарию, но и более редкие формы патологического состояния. Иногда его устанавливают при волосатоклеточным лейкозе, лимфатическом, в качестве осложнения которого – цитолиз. Известно, что комплекс симптомов может развиться на фоне медикаментозной терапии, облучения, влияния химических компонентов. Большое внимание в современной медицине привлекает посттрансплантационный лимфопролиферативный синдром, существенно ухудшающий прогнозы перенесшего операцию человека. В развитии синдрома, полученного не наследственным путем, наиболее сильно влияние ретровирусов.
Нюансы и распространенность
Медицинская статистика показывает, что преимущественный процент пациентов с лимфопролиферативным синдромом – люди старше пятидесятилетнего возраста. Изредка заболевание выявляется у тех, кто младше 25, но такие случаи единичны. Среди мужского пола частота встречаемости в среднем вдвое выше, нежели у женщин. Исходя из течения, говорят о доброкачественной форме, спленомегалической, опухолевой, склонной к быстрому прогрессу, затрагивающей костный мозг, брюшную полость. Также есть пролимфоцитарный тип.
Когда только начинает развиваться лимфопролиферативный синдром, внутренние болезни не беспокоят, человек чувствует себя удовлетворительно, отсутствуют активные жалобы. Некоторые отмечают слабость, склонность к простудам. Несколько активнее нормы функционируют потовые железы. Заболевание на этом этапе можно выявить в рамках профилактического обследования или на случайном осмотре. Основные признаки – ненормально крупные лимфоузлы, лимфоцитоз, повышение концентрации лейкоцитов в кровеносной системе.
Специфика симптоматики
При заболевании склонны к увеличению лимфоузлы на шее, в подмышечной ямке. Несколько позже, когда болезнь приобретает развернутую форму, отмечается увеличение прочих групп. Размеры сильно варьируются, как и консистенция: некоторых похожи на неплотное тесто, при исследовании болью не отзываются, между собой или с кожными покровами не сливаются. Для таких участков нехарактерно формирование язв или нагноений.
Когда болезнь приобретает развернутую форму, проявления становятся ярко выраженными, пациент ощущает себя слабым, резко понижается способность работать. У больного активно работают потовые железы, он теряет вес, страдает от жара. Лимфатические узлы существенно увеличены, что и привлекает внимание при первичном осмотре.
Осмотр больного: комплекс проявлений
При обследовании пациента заподозрить лимфопролиферативный синдром можно, если четко диагностируется лифмоаденопатия. У многих больных видна трансформация отдельных участков кожи: появляется инфильтрат, выявляются неспецифические пораженные участки. Если человек ранее страдал от кожных заболеваний, они обостряются в силу описываемого синдрома. Многих беспокоит эксфолиативная эритродермия. На фоне синдрома возможно развитие герпеса, крапивницы, дермита.
Для уточнения состояния необходимо направить больного на КТ, УЗИ. На лимфопролиферативный синдром указывает рост лимфоузлов в грудине, брюшной полости, при этом состояние не всегда сопровождается проявлениями компрессии. У пациента больше нормы селезенка, печень. Изучение слизистых пищеварительного тракта позволяет заметить лейкемическую инфильтрацию. Дополнительные проявления – язвы в желудке, кишечном тракте, кровотечения в этой области. Есть вероятность мальабсорбционного синдрома.
Прогресс состояния
При лимфопролиферативном синдроме возможно вовлечение дыхательной системы в патологические процессы. Лейкемическая инфильтрация может затронуть как верхние отделы, так и нижние пути прохождения воздуха. Больной кашляет, беспокоит одышка, возможно отхаркивание мокроты с кровянистыми включениями. Иногда устанавливают плеврит.
В ряде случаев описанный синдром провоцирует инфильтрацию почечной паренхимы. Такое состояние крайне редко проявляет себя типичной симптоматикой. Возможно распространение инфильтрата на ЦНС, что приводит к менингиту, некоторым формам энцефалита и параличу нервных структур, может стать причиной комы. При распространении инфильтрата на кавернозные тела больной страдает от продолжительной и провоцирующей боль эрекции, в медицине называемой приапизмом.
Лабораторные анализы
При подозрении на лимфопролиферативный синдром пациента направляют на исследование крови. Указанное состояние сопровождается ростом концентрации лимфоцитов, лейкоцитов. Возможна анемия.
Лабораторные анализы помогают диагностировать у пациента гемат-, протеинурию. Анализ на биохимию уточняет гипогаммаглобулинемию. В небольшом проценте случаев у пациентов устанавливают гипоальбуминемию. Гепатоцитный цитолиз указывает на себя гиперферментемией.
Иммунологическое исследование указывает на повышение концентрации в селезенке, кровеносной системе лимфоцитов, сбой баланса хелперов и супрессоров из числа лимфоцитов. Вместе с тем снижается концентрация IgG, IgA, IgM (для двух последних изменения особенно ярко выражены). Иммунофенотипирование – основание заключить, что лейкозные клеточные структуры – CD 5, 19, 20, 23 из класса В-лимфоцитов. Результаты цитогенетического анализа в 65 % случаев указывают на аномалии хромосом.

Что делать?
При лимфопролиферативном синдроме больному показано соблюдение разработанного врачом лечебного режима – программа выбирается индивидуально. Пациенту назначают цитостатические препараты. Особенно актуально это, если состояние здоровья быстро ухудшается, печень и селезенка, лимфоузлы стремительно увеличиваются. Цитостатики незаменимы при лейкемической инфильтрации волокон ЦНС, а также в случае, если процессы затрагивают органы вне кроветворной системы. Состояние указывает на себя сильной болью и сбоем функциональности систем и органов.
Если количество лейкоцитов в кровеносной системе неуклонно и быстро растет, показаны хлорбутин, спиробромин. Неплохую реакцию организма позволяют получить проспидин, циклофосфан. Иногда врачи рекомендуют остановиться на пафенциле. Если к этому есть специфические показания, могут назначить полихимиотерапию. В рамках такого курса цитостатические средства, влияющие на организм разными образами, комбинируют между собой.
Мероприятия и способы: как помочь пациенту?
При повышении содержания лейкоцитов до уровня 200*10 на 9/л рекомендован лимфоцитафарез. Если отдельные лимфоузлы резко и сильно увеличиваются, такие процессы выявлены в селезенке, если лимфаденопатия переходит в системную генерализованную форму, назначают лучевое лечение. При разрастании селезенки, не корректирующемся медикаментозными средствами и облучением, пациенту рекомендована спленэктомия. Ее необходимо пройти, если часты инфаркты этого органа, а также при заболевании, сопровождающемся выраженной спленомегалией, определенными формами лейкоцитозома, лейкоза. Спленэктомия незаменима при гранулоцито-, эритро-, тромбоцитопении, анемии аутоиммунного типа, тромбоцитопении, которую не удается регулировать глюкокортикоидами.
Если гормональные соединения показывают выраженный эффект при тромбоцитопении, если установлена гемолитическая анемия, а предваряющим для этого патологического состояния был хронический лимфолейкоз, назначают глюкокортикоиды как основной курс терапии. Эти препараты помогают при хроническом сублейкемическом течении лимфолейкоза, сопровождающемся сильным разрастанием печени, селезенки, лимфатических узлов. Глюкокортикоиды используют, если пациент не переносит цитостатические медикаменты, нет возможно применить облучение либо патологическое состояние проявляет стойкость к таким терапевтическим подходам.
Важные нюансы
Гормональные средства незаменимы, если цитостатические стали причиной цитопении, геморрагического синдрома. Их применяют в рамках полихимиотерапии, комбинируя основной курс и преднизолон.
Для описываемого патологического состояния характерны осложнения инфекционной природы. При таком развитии ситуации больному показан курс антибиотиков. Чаще всего применяют препараты обширного спектра эффективности. Хорошо зарекомендовали себя макролиды, аминогликозиды. Можно использовать полусинтетические средства из пенициллинового ряда, цефалоспоринового, иммуноглобулин.
Миелопролиферативный синдром
Это патологическое состояние нередко рассматривают в рамках образовательной программы вместе с описанным выше. Термином принято обозначать патологию, при которой активно вырабатываются миелоидные клетки. Причина явления – неправильная работа стволовых клеток системы, ответственной за производство крови. Синдром объединяет в себя несколько болезней – лейкоз, миелофиброз, тромбоцитоз, полицитемию. Сюда же принято относить миелодиспластический синдром.
Хронические лимфопролиферативные заболевания (хЛПЗ) - группа клональных неопластических заболеваний лимфопоэтической системы с клеточной пролиферацией В- ,Т- или NK-лимфоцитов на различных уровнях их дифференцировки.
Чаще всего эти заболевания поражают лиц пожилого возраста, протекают хронически с медленным прогрессированием и выраженным иммунодефицитом.
С учетом специфических клинических, морфологических, иммунофенотипических и цитогенетических данных целесообразно выделение в рамках хЛПЗ следующих подгрупп:
- неходжкинские лимфомы,
- лимфома Ходжкина - лимфогранулематоз,
- моноклональные гаммапатии.
Лимфома Беркитта
Лимфома Беркитта (ЛБ) - В-клеточная лимфома высокой степени злокачественности с наличием ряда специфических характеристик:
- отчетливая связь с инфицированием вирусом Эпштейн-Барра,
- географические особенности распространения (эндемический вариант в 80% встречается в Африке, Новой Гвинее; спорадический тип в 20% - в Европе и США),
- t (8; 14) (q24; q32) с реаранжировкой онкогена MYC,
- экспрессия антигенов CD19, CD20, CD22, CD10, CD43, CD79a при отсутствии экспрессии CD5, CD23, sIgM, BCL-2.
При эндемическом варианте лимфома поражает кости лицевого скелета с поражением зрительного и лицевого нервов. В 20% случаев поражается костный мозг (что расценивается как ОЛЛ при количестве бластов более 20%), часто вовлекаются в процесс менингеальные оболочки с развитием картины нейролейкемии.
Спорадическому типу присуще поражение лимфоузлов брюшной полости и тонкой кишки с вторичным вовлечением ретроперитонеальных структур (почки, поджелудочная железа), хотя некоторые авторы считают, что поражение почек и ретроперитонеального пространства характерно и для эндемического варианта лимфомы Беркитта.
Учитывая высокоагрессивный характер лимфомы Беркитта, необходимо проведение активной полихимиотерапии (ПХТ) с обязательной профилактикой поражения ЦНС. Рекомендуется выполнение протоколов ПХТ Hyper-CVAD, CODOX-M/IVAC. Гематологический центр РАМН рекомендует лечение ЛБ по протоколу ЛБ-М-04 продолжительностью 4 месяца (дизайн приводится ниже).
циклофосфамид 200 мг/м2 в/венно капельно 1-5 дни,
дексаметазон 10 мг/м2 в/венно капельно 1-5 дни.
• дексаметазон 10 мг/м2 в/венно 1-5 дни,
• ифосфамид 800 мг/м2 в/венно 1-5 дни,
• метотрексат 1,0 г/м в/венно 1-й день, 12ч,
• доксорубицин 50 мг/м2 в/венно 3-й день,
• цитарабин 150 мг/м2 х 2 в день 4-5 дни,
• винкристин 2 мг в/венно, 1-й день,
• вепезид 120 мг/м2 в/венно, 4-5 дни,
• пункция с введением трех препаратов
• интратекально: цитарабин 30 мг,
• метотрексат 15 мг, преднизолон 30 мг.
• дексаметазон 10 мг/м2 в/венно 1-5 дни,
• метотрексат 1,0 г/м2 в/венно 1-й день, 12 час,
• винбластин 10 мг в/венно струйно, 1-й день,
• цитарабин 2,0 г/м2 в/венно х 2 в день 2-3 дни
• вепезид 150 мг/м2 в/венно 3-5 дни, пункция с введением трех препаратов интратекально: цитарабин 30 мг, метотрексат 15 мг, преднизолон 30 мг.
Проводится 4 блока полихимиотерапии по схеме А-С-А-С с интервалами между блоками в 21 день, начиная от первого дня предыдущего курса. Сопроводительная терапия проводится так же, как и при ПХТ ДВККЛ (см. предыдущий протокол терапии).
Пролимфоцитарный хронический лимфолейкоз
Для пролимфоцитарного лейкоза характерно наличие спленомегалии, умеренной лимфоаденопатии, высокого лейкоцитоза. В крови и миелограмме преобладают пролимфоциты - крупные клетки с большим круглым ядром и отчетливо видимой нуклеолой. Цитогенетически нередко можно выявить аномалию 14q- при В-клеточном и часто - изменения Хр11 и гена ATM при Т-клеточном ПЛЛ.
Пролимфоцитарный лейкоз отличается агрессивным течением и недостаточным эффектом от применяемых в лечении ХЛЛ протоколов. Длительность жизни больных при ПЛЛ составляет 2-3 года.
Обычно в лечении пролимфоцитарного лейкоза применяют интенсивную полихимиотерапию, в т.ч. в сочетании с аналогами пуриновых нуклеозидов или моноклональными антителами (CHOP, R-CHOP, FMD, FCR). Применение флюдарабина и пентостатина в комплексной ПХТ позволяет достичь ремиссии у 50% больных. Возможно также выполнение спленэктомии или облучение селезенки с целью уменьшения массы опухоли.
Т-клеточный хронический лимфолейкоз
Согласно классификации ВОЗ (2001 г.), данное заболевание является Т-клеточным ХЛЛ/Т-клеточным ПЛЛ и относится к периферическим опухолям Т-клеток. Т-клеточный фенотип встречается в 2-3% случаев хронического лимфолейкоза, обычно заболевают люди молодого возраста.
У большинства больных наблюдается спленомегалия, нередко в сочетании с гепатомегалией; лимфоаденопатия бывает относительно редко; часто имеются поражения кожи вследствие ее лейкемической инфильтрации.
В анализах периферической крови обнаруживается высокий лейкоцитоз с наличием 30-10% зрелых лимфоцитов и более 20% пролимфоцитов. Клетки опухоли имеют фенотип CD3+, CD4-, CD8-, однако при вялотекущем варианте может быть экспрессия CD8. Очень редко выявляются аномалии Хр8 и Хр14. Болезнь протекает со слабой чувствительностью к алкилирующим агентам, однако достаточно эффективно лечение по протоколам с включением аналогов пуриновых нуклеозидов.
Другим вариантом Т-ХЛЛ является ХЛЛ из больших гранулярных лимфоцитов. Его подразделяют на два подтипа: лейкоз с иммунофенотипом Т-лимфоцитов - CD8+, CD4- CD56- и лейкоз из Т-лимфоцитов с иммунофенотипом NK(CD56+). Для первого подтипа характерно медленное течение и скудная симптоматика.
Больные жалуются на быструю утомляемость и общую слабость; лимфоаденопатия и гепатомегалия встречаются крайне редко, спленомегалия выявляется не более чем у 20% больных. У некоторых больных имеет место интоксикационный синдром, довольно часто развиваются аутоиммунная гемолитическая анемия (АИГА), реактивные артриты. В анализах крови отмечается анемия, гранулоцитопения.
Опухолевые клетки крупнее, чем зрелые лимфоциты, содержат круглое или овальное ядро, расположенное эксцентрично. Цитоплазма обильная бледно-голубая с наличием азурофильных гранул. У большинства больных обнаруживается диффузная лимфоцитарная инфильтрация костного мозга.
Второй подтип чаще встречается у лиц молодого возраста, одинаково часто у мужчин и женщин. При нем часто отмечается интоксикационный синдром. Типично наличие выраженной гепатоспленомегалии при отсутствии лимфоаденопатии, иногда бывает поражение желудочно-кишечного тракта.
В анализах крови отмечается высокий лейкоцитоз, умеренная гранулоцитопения. Заболевание отличается агрессивным течением и слабой реакцией на терапию. Умеренный эффект отмечен от терапии по протоколам с включением аналогов пуриновых нуклеозидов.
Общие нарушения в организме при лейкозах проявляются в виде ряда клинических синдромов: анемического, геморрагиче- ского, инфекционного, интоксикационного, пролиферативного (метастатического).
Анемический синдромсвязан c подавлением эритропоэза вследствие дисплазии, или вытеснения нормального эритроидно- го ростка из костного мозга лейкозным, что приводит к развитию гипо- или апластической анемии. Другими причинами являются нарушение усвоения витамина В12 и железа эритробластами, ге- молиз эритроцитов. У больных появляется бледность, одышка, сердцебиение.
Рис. 55. Геморрагии на слизистых оболочках ротовой полости при остром лейкозе
Рис. 56. Гипертрофия десен и геморрагии при остром монобластном лейкозе
Инфекционный синдромобусловлен неспособностью лей- козных клеток к выполнению защитных функций (фагоцитоз, специфическая иммунологическая реактивность) вследствие кле- точного атипизма), а также лейкопеническим синдромом. Вслед- ствие этих причин организм больного лейкозом становится легко уязвимым не только для патогенной микрофлоры, но и для ус- ловно-патогенных микроорганизмов. У пациентов выявляются как легкие (локальные) формы инфекций (кандидозные стомати- ты, гингивиты, поражения слизистых оболочек), так и тяжелые генерализованные процессы (пневмонии, сепсис).
Пролиферативный (метастатический) синдром:
§поражение других органов и тканей.
Лимфоаденопатия.Происходит увеличение любой группы лимфатических узлов в связи с пролиферацией в них лейкозных лимфоидных клеток. При этом выявляются множественные, плотные, эластичные, округлые конгломераты лимфоузлов, кото- рые могут быть спаяны друг с другом, размером от 1 до 8 см; при пальпации они безболезненны.
Увеличение брыжеечных лимфатических узлов и гипертро- фия червеобразного отростка (как лимфоидного органа) могут вызывать боль в области живота. Гипертрофированные внутри- грудные лимфатические узлы могут привести к сдавлению средо- стения.
Печень и селезенка также увеличены (рис. 57). Увеличение
их размеров связано с метастазированием в эти органы лейкоз-
ных клеток и образованием в них экстрамедуллярных очагов ге-
Рис. 57. Увеличение селезенки при метастатическом синдроме
Нейролейкоз. При лейкозах возможно паражение ЦНС. Наиболее часто возникает при остром лимфолейкозе и значи- тельно ухудшает течение и прогноз. Возникновение нейролейко- за обусловлено метастазированием лейкозных клеток в оболочки головного и спинного мозга или в вещество мозга. В результате развиваются различной степени тяжести нарушения неврологи-
ческого статуса – от легкой общемозговой симптоматики (голов- ная боль, головокружение) до тяжелых очаговых поражений (на- рушение сознания, снижение остроты зрения, дискоординация движений, дисфазия).
Возможно развитие специфических узелков – лейкемидов кожи, метастазирование в вилочковую железу с гипертрофией тимуса.
Рис. 58. Очаги просветления при радиографии черепа при множественной миеломе
Интоксикационный синдромсвязан с повышением в кро- ви нуклеопротеидов – токсических продуктов, образующихся при распаде (гибели) лейкозных клеток. Проявляется лихорадочным и болевым синдромами, снижением аппетита, массы тела, общей слабостью.
Хронические лейкозы необходимо дифференцировать с
Лейкемоидные реакции
Под лейкемоидными реакциями(лейкемия + eides – по- добный) понимают патологические реакции системы крови, сходные с лейкозами по картине периферической крови (увели- чением лейкоцитов, появлением незрелых форм лейкоцитов), но отличающиеся от них по патогенезу.
Чаще они характеризуются высоким лейкоцитозом с резко выраженным сдвигом лейкоцитарной формулы влево и появле- нием молодых форм гранулоцитов вплоть до единичных бластов. Однако, иногда возможны лейкемоидные реакции проявляющие- ся лейкопенией.
Лейкемоидные реакции являются одним из симптомов дру-
гих заболеваний и возникают в ответ на внедрение в организм
агентов биологической природы (вирусов, риккетсий, микроорга-
низмов, паразитов), на действие биологически активных веществ,
высвобождающихся при иммунных и аллергических процессах,
при распаде тканей.
В отличие от лейкозов, для которых характерна злокачест-
венная трансформация кроветворных клеток, механизм развития
лейкемоидных реакций заключается в реактивной очаговой ги-
перплазии различных нормальных ростков лейкопоэтической
ткани и выхода в кровь большого количества незрелых лейкоци-
тов, включая их бластные формы. После купирования первичного
заболевания, вызвавшего лейкемоидную реакцию, патологиче-
ские изменения в крови исчезают.
К лейкемоидным относятся реакции миелоидного и лимфо- цитарного типа. В свою очередь, лейкемоидные реакции миело- идного типа подразделяют на реакции с картиной крови, соответ- ствующей хроническому миелолейкозу (тяжелые инфекционно- воспалительные процессы, интоксикации, лимфогранулематоз), миелобластного (сепсис, туберкулез) и эозинофильного типа (па- разитарные инвазии, аллергические заболевания, коллагенозы).
Среди лейкемоидных реакций лимфоцитарного типа выде- ляют монолимфоцитарные (инфекционный мононуклеоз), по кар- тине крови напоминающие хронический лимфолейкоз, и лимфа- тического типа с гиперлейкоцитозом, которые часто наблюдается
на фоне вирусных инфекций. Преходящий характер реакции лимфатического типа, возникновение только в детском возрасте отличает ее от хронического лимфолейкоза.
Таблица 15. Отличия лейкемоидных реакций от лейкозов
| Лейкемоидные реакции | Лейкозы | |
| Проявляется после перенесенной инфекцией | да | нет |
| Купирование инфекции приводит к нормализации картины крови | да | нет |
| Наличие анемии и тромбоцитопении | нет | да |
| Наличие токсической зернистости нейтрофилов | да | нет |
Диагностика лейкозов
Диагностика лейкозов может осуществляться при использо- вании различных методов. Выделяют следующие способы диаг- ностики лейкозов:
§ гематологическое исследование (анализ крови, пункта-
та костного мозга);
§ гистологическое исследование крыла подвздошной
§ иммунологическое исследование (с помощью флюо-
ресциирующих моноклональных антител);
§ цитогенетическое исследование (например, выявление
филадельфийской хромосомы при хроническом миелолейкозе);
§ цитохимическое исследование (выявление кислой фосфатазы, миелопероксидазы, эстеразы и других ферментов).
При анализе крови подозрение на лейкоз может возникнуть при налии клинических симптомов и изменений в перифериче- ской крови: присутствие бластов, анемия, изменение количества лейкоцитов, нейтропения, лимфоцитоз, тромбоцитопения.
Пункция костного мозга. При многих заболеваниях крове- творных органов изучение клеточного состава костного мозга имеет большое диагностическое значение и служит важным до- полнением к результатам, полученным при исследовании гисто- логического состава крови (приложение 1, 2).
Значение его заключается в том, что дегенеративные изме-
нения форменных элементов крови, усиленная регенерация, раз-
личного рода нарушення нормального эритропоэза, лейкопоэза
нередко в костном мозгу проявляются раньше и в более ясной
форме, чем в крови. После просмотра мазков подсчитывается
миелограмма– процентное содержание различных клеток кост-
ного мозга. Вычисляется лейко-эритробластическое отношение –
отношение суммы клеток лейкоцитарного ряда к сумме клеток
эритроцитарного ряда. В норме оно равно 2,5:1- 4:1. При лейко-
зах лейко-эритробластическое отношение увеличивается.
Методика стернальной пункции
Пункцию проводят в процедурном кабинете с соблюдением правил асептики. Используют иглу Кассирского с предохрани- тельным щитком-ограничителем и шприц на 10-20 мл. Игла и шприц должны быть стерильны и высушены спиртом и эфиром.
У взрослых пунктируют чаще грудину (рукоятку или на уровне третьего-четвертого межреберья) по средней линии. Можно пунктировать подвздошную кость, ребра. У детей пунк- тируют подвздошную кость, пяточную, нижний эпифиз бедрен- ной кости, грудину.
Иглу вводят быстрым движением в костно-мозговой канал.
После извлечения мандрена на иглу насаживают шприц и аспи-
рируют костный мозг. Полученный материал переносят на стекло
с луночкой или часовое стекло. Пунктат слегка перемешивают
стеклянной палочкой. Учитывая быструю коагуляцию пунктата, все дальнейшие манипуляции делают быстро. Из капель пунктата готовят тонкие мазки обычным образом.
В редких случаях производится пункция лимфатического узла. Микроскопическое исследование приготовленного из пунк- тата и окрашенного обычным способом мазка позволяет иногда получить более детальное представление о характере кроветво- рения в лимфатической системе.
Следует отметить, что острый лейкоз наиболее часто проте-
кает на фоне умеренного лейкоцитоза или лейкопении, тогда как
хронический миело- и лимфолейкоз обычно сопровождаются
очень высоким лейкоцитозом. При анализе миелограммы у боль-
ных с лейкозами обращает на себя внимание увеличение содер-
жания бластных форм более 5% с различной морфологией, лим-
фоцитозом, отсутствием мегакариоцитов (за исключением остро-
го мегакариобластного лейкоза).
Иммунологическая диагностика (иммунофенотипирова- ние бластов). В диагностике лейкоза на современном этапе большое значение имеет метод проточной цитометрии. Это авто- матизированная методика, суть которой состоит в том, что клетки крови обрабатывают моноклональными антителами с присоеде- ненной флюоресцентной меткой и направляют с потоком жидко- сти в капилляр, освещенный лазером. Измерение интенсивности флюоресценции отдельных клеток, проходящих через лазерный пучок, позволяет оценить количество экспрессируемых ими ан- тигенов (СD-антигенов или кластеров дифференцировки). Этот метод позволяет точно диагностировать тип лейкоза, что важно для определения схем лечения.

Рис. 59. Иммунологическая диагностика лейкозов
Наличие светящегося ореола вокруг лейкоцитов – следствие взаимодействия моноклонального антитела, меченного флюоресцирующей меткой, с антигеном, к которому данное антитело специфично
Цитогенетическая диагностикапозволяет выявить геном- ные и хромосомные мутации – изменение количества хромосом (транслокации, делеции и др.) либо их качества. Хромосомные аномалии отмечаются у 80-90% пациентов с острым миелобласт- ным лейкозом и хроническим миелолейкозом и у 50% больных – с хроническим лимфолейкозом.
Цитохимическая диагностиказаключается в определении
специфических для различных видов лейкозных клеток фермен-
тов и включений.
|