
Лимфоматоидный папулез что это



Лимфоматоидный папулез
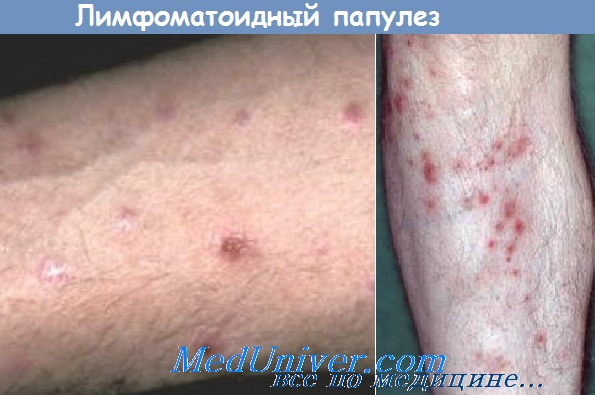
Лимфоматоидный папулез относится к заболеваниям лимфатической системы, проявляется рецидивирующими полиморфными высыпаниями. Высыпания множественные, больных не беспокоят, разрешаются самостоятельно. При гистологическом исследовании находят лимфоциты с признаками атипизма. Возможно злокачественное перерождение, хотя риск и невысок.
Синоним: papulosis lymphomatoides .
Эпидемиология
1,2-1,9 на 1000 000.
Любой; средний возраст больных — 40 лет.
Мужчины и женщины болеют одинаково часто.
Не играет никакой роли.
Сыпь обычно не беспокоит больных, хотя иногда отмечаются зуд, боль, болезненность. Для лимфоматоидного папулеза нехарактерны похудание, потеря аппетита, потливость и лихорадка. При появлении подобных симптомов надо заподозрить лимфому.
Сыпь появляется волнами и исчезает самостоятельно. Элемент сыпи может разрешиться на любой стадии развития, полный цикл которого занимает от 2 до 8 нед. Рубцы остаются только на месте самых старых элементов.
Элементы сыпи. Свежие высыпания представлены папулами (рис. 21-23), узлами, изредка — опухолевидными образованиями. Сначала поверхность элементов гладкая; они часто имеют геморрагический компонент. Гиперкератоз приводит к появлению чешуек. С течением времени центр элемента нек-ротизируется и покрывается коркой, возможно изъязвление (рис. 21-24). Диаметр элементов — от 2 до 30 мм, количество — от двух-трех до нескольких сотен. После заживления остаются атрофические рубцы, иногда с послевоспалительной гипер- или гипопигментацией.
Цвет. От красного до бурого. Центральные некротизированные участки черного цвета. Расположение. Беспорядочное или в виде групп. У некоторых больных элементы образуются вокруг волосяных фолликулов. Локализация. В основном туловище и конечности. Изредка — слизистая рта и половых органов.
Дифференциальный диагноз
Множественные папулы или узлы на разных стадиях развития
Острый лихеноидный оспенновидный па-рапсориаз (парапсориаз типа Мухи—Ха-бермана, в гистологических препаратах отсутствуют атипичные лимфоциты). Неход-жкинские лимфомы кожи и лимфогранулематоз (неуклонно прогрессируют и имеют иную гистологическую картину). Анапла-стическая Т-крупноклеточная лимфома, атипичный регрессирующий гистиоцитоз и грибовидный микоз (более крупные папулы, инфильтраты не имеют клиновидной формы, нет васкулита и почти нет диапеде-за эритроцитов, сильнее выражен экзоци-тоз). Доброкачественная лимфоплазия, гистиоцитоз X , лекарственная токсидермия, папулезная крапивница, чесотка, вирусные инфекции.
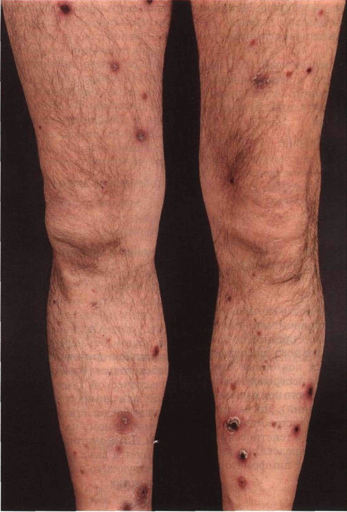
Рисунок 21-23. Лимфоматоидный папулез. Множество красновато-бурых папул и узлов; в центре элементов — кровоизлияния, некрозы и плотные корки ные. Многополюсные митозы. Атипичные лимфоциты напоминают клетки Рид-Штернберга, характерные для лимфогранулематоза.
Лимфоматоидный папулез типа В. Мелкие атипичные лимфоциты с церебриформным ядром. Митозов немного. Атипичные лимфоциты напоминают клетки Лутцнера при грибовидном микозе.
Атипичные клетки несут маркеры активированных Т-лимфоцитов: рецептор интер-лейкина-2 ( CD 25), HLA - DR , Ki -1 ( CD 30), CD 4. Блоттинг по Саузерну: в 50% случаев — пролиферация одного клона Т-лимфоцитов.
Внутренние органы не поражены.
Типичная гистологическая картина, результаты иммунофенотипирования, отсутствие поражения внутренних органов по данным анамнеза и физикального исследования. Могут потребоваться повторные биопсии кожи для исключения перерождения в лим-фому (наиболее вероятного при наличии опухолевидных образований). До настоящего времени нет надежных гистологических, им-муногистохимических и генетических критериев, позволяющих предсказать злокачественное перерождение лимфоматоидного папулеза.
Этиология и патогенез
Неизвестны. Одни рассматривают лимфоматоидный папулез как лимфому низкой степени злокачественности, рост которой сдерживается иммунной системой, другие — как локализованную доброкачественную лимфоплазию с участием лимфоцитов CD 4. Одинаковые маркеры атипичных лимфоцитов и возможность трансформации лимфоматоидного папулеза в лимфогранулематоз и Т-клеточные лимфомы кожи подтверждают первую точку зрения, а редкость таких наблюдений — вторую. Поликлональная (ре- активная) лимфоидная пролиферация при лимфоматоидном папулезе может выйти из-под контроля иммунной системы и стать моноклональной (опухолевой), что приведет к развитию злокачественной лимфомы. Лимфоматоидный папулез вместе с атипичным регрессирующим гистиоцитозом, ана-пластической Т-крупноклеточной лимфо-мой и лимфогранулематозом относятся к первичным лимфопролиферативным заболеваниям кожи, при которых атипичные клетки экспрессируют антиген Ki -1 ( CD 30).
Клиническое значение
Заболевание хроническое, возможно злокачественное перерождение лимфоматоидного папулеза.
Течение и прогноз
Течение болезни бывает разным. У одних больных высыпания разрешаются через 3 нед, у других сохраняются десятилетиями; у одних наблюдаются длительные ремиссии, у других волны высыпаний следуют друг за другом без перерыва. 10—20% больных страдают и другим лимфопролиферативным заболеванием (грибовидным микозом, ана-пластической Т-крупноклеточной лимфо-мой или лимфогранулематозом), которое может предшествовать, сопутствовать лим-фоматоидному папулезу либо следовать за ним. Лимфоматоидный папулез нередко сохраняется даже на фоне химиотерапии сопутствующего лимфопролиферативного заболевания.
Ни один из существующих методов лечения нельзя признать радикальным.
Применяют местное лечение кортикосте-роидами и кармустином, облучение электронным пучком. PUVA -терапия эффективна, но на долгосрочный прогноз не влияет. Сообщалось об эффективности тетрацикли-нов, дапсона и приема кортикостероидов внутрь. В некоторых клиниках используют ретиноиды, метотрексат, хлорамбуцил, циклоспорин, циклофосфамид и интерферон а-2Ь.
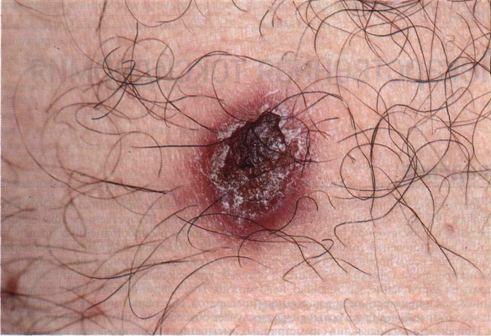
Рисунок 21-24. Лимфоматоидный папулез. Типичный красновато-бурый узел; некро-тизированный центральный участок покрыт коркой

Лимфоматоидный папулез (болезнь Маколея) представляет собой доброкачественную кожную патологию, связанную с лимфатической системой, имеющую рецидивирующие полиморфные проявления.
Характерные поражения патологии – это различные по размеру папулезные высыпания, которые впоследствии преобразовываются в буллы. В дальнейшем они вскрываются, и на их месте образовываются эрозии, корочки и рубцы.
Проявления могут возникать по всему телу в единичном экземпляре или во множественном числе.
При постановке диагноза дерматологи учитывают анамнез пациента и характер проявлений. Дополнительно делается гистологическое и иммуногистохимическое исследование.
Для лечения этой кожной патологии назначают глюкокортикостероидные препараты, цитостатики, антибактериальные средства и метод ПУВА-терапии. Но все предпринятые меры помогают только кратковременно.
Средний возраст дебюта лимфоматоидного папулеза – 40 лет.
Классификация патологии проводится по гистологической картине, на основании этого дерматологи выделяют четыре разные формы лимфоматоидного папулеза:
- Тип А – гистологические данные выявляют различные виды клеток (лимфоциты, нейтрофилы, эозинофилы, гистиоциты и анапластические лимфоидные клетки);
- Тип В – редко встречающаяся форма заболевания, по своему проявлению похожа на грибовидный микоз. При исследовании обнаруживаются атипичные единичные клетки лимфомы.
- Тип С – напоминает крупноклеточную лимфому, в препарате преобладают злокачественные лимфоидные клетки;
- Тип D – клиническая картина схожа с заболеванием педжетоидный ретикулёз. Является наиболее агрессивной из всех форм.
Симптомы
Патологический процесс начинается с высыпаний на эпидермисе (в редких случаях сначала могут поражаться слизистые). Проявления возникают в виде пятен бурого цвета, в диаметре вырастающих до 3 см, которые могут слегка зудеть.
На протяжении нескольких недель на их месте образовываются узелки, опухоли, буллы, которые сверху покрывают чешуйки. Высыпания появляются на любом участке тела, на лице, на коже головы, редко – на слизистых.
Патологические элементы высыпают волнообразно, способны саморазрешаться. Также имеют склонность к образованию групп, особенно часто скапливаются вокруг волосяных мешочков.
Весь цикл существования патологических элементов при этом заболевании может составлять от 2х недель до 2х месяцев. В течение этого промежутка узлы вскрываются, при этом появляются зудящие эрозии, которые покрываются корочками.
Кроме этого, возникают расчёсы, и присоединяется инфекция. В некоторых случаях появляется шелушение. Все эти проявления создают картину ложного полиморфизма. Любой патологический элемент может инволютировать в разной фазе развития.
После разрешения первичных элементов образуется рубец, на месте которого остается гипер- или гипопигментация.
Причины
Этиология и патогенез патологии до сих пор не изучены. Большинство специалистов, опираясь на гистологическую картину этой болезни, рассматривают ее как, лимфоцитарную опухоль кожного покрова, имеющую низкую степень злокачественности.
Другая часть дерматологов считают, что лимфоматоидный папулез – это ограниченная доброкачественная лимфоплазия, которая появляется при участии CD4-лимфоцитов, регулирующих иммунитет.
Заболевание развивается из-за нарушений в дифференцировке Т-лимфоцитов, когда они начинают мутировать.
Таким образом провоцируется усиленная пролиферация эпидермальных клеток, не только здоровых, но и с генетическим сбоем.
Параллельно меняются внутренние функции клеток эпидермиса, которые начинают производить цитокины, а также медиаторы, включающие механизм воспаления и усиливают пролиферацию клеток измененной кожи. Так образовываются доброкачественные лимфоидные элементы дермы.
Экспрессия CD30-антигена нетипичными клетками, сочетающаяся с возможностью неожиданной трансформации лимфоматоидного папулеза в недоброкачественную лимфому свидетельствуют в пользу теории о том, что эта патология является одним из видов опухоли, имеющих небольшую степень злокачественности.
Однако такая трансформация крайне редко происходит, поэтому теория лимфоплазии ближе к истине.
Доподлинно известно, что если пролиферация лимфоцитов выйдет из-под контроля иммунной системы, то образуется злокачественная лимфома.
Диагностика и лечение
Диагноз лимфоматоидного папулеза ставится на основании анамнеза, клинических кожных поражений, данных гистологии и иммуногистохимического исследования. Иногда для изучения берут ткань измененного лимфатического узла, который расположен поблизости.
В спорных случаях выполняют цитогенетическое и молекулярное исследование.
Дифференцируют эту патологию с другими кожными заболеваниями, такими как: крапивница, чесотка, парапсориаз, синдром Сезари и другими.
Радикальные методы терапии лимфоматоидного папулеза не придуманы. Лечение проводят с помощью больших доз тетрациклинов, которые совмещают с кортикостероидами, цитостатиками и сульфонами. В редких случаях добавляют ретиноиды и интерфероны.
Диссеминированные проявления облучаются электронным пучком, дополнительно проводится ПУВА-терапия. Все применяемые препараты и методы не дают долгосрочного эффекта.
Прогноз напрямую зависит от течения заболевания и количества рецидивов.
Профилактика
Из-за невыясненной этиологии не существует профилактических мер заболевания. Больным рекомендуется регулярно наблюдаться у дерматолога, чтобы как можно раньше заметить рецидив и начать его лечение.
Лимфоматоидный папулез – доброкачественное заболевание лимфатической системы с рецидивирующими полиморфными кожными проявлениями. Клинически сопровождается папулезными высыпаниями различной величины, которые затем трансформируются в буллы, вскрываются, образуя эрозии, корочки, рубцы. Сыпь может располагаться на любом участке кожного покрова, субъективные ощущения отсутствуют. Диагноз выставляется на основании анамнеза, клинических данных, гистологического и иммуногистохимического исследований. В рамках лечебного курса лимфоматоидного папулеза применяются глюкокортикостероиды, антибиотики, цитостатики, ПУВА-терапия, однако все меры дают лишь кратковременный эффект.
МКБ-10
- Причины лимфоматоидного папулеза
- Классификация лимфоматоидного папулеза
- Симптомы лимфоматоидного папулеза
- Диагностика и лечение лимфоматоидного папулеза
- Цены на лечение
Общие сведения
Лимфоматоидный папулез – узелковая лимфоидная опухоль кожи доброкачественного характера, имеющая тенденцию к медленному росту. Может быть представлена как единичным образованием, так и множественными элементами. Заболевание не имеет гендерной окраски, сезонности, эндемичности, наследственной предрасположенности. Средний возраст манифестации патологии - 40 лет. Распространенность в популяции составляет 1-2 случая на 1 млн. человек. У представителей негроидной расы лимфоматоидный папулез практически не встречается. В самостоятельную нозологическую единицу заболевание выделил В. Маколей в 1968 году, отсюда другое название лимфоматоидного папулеза – болезнь Маколея.
В настоящее время лимфоматоидный папулез принято относить к группе первичных CD30+ лимфопролиферативных патологий кожи, наряду с такими процессами, как псевдолимфома, лимфогранулематоз, атипичный регрессирующий гистиоцитоз, анапластическая крупноклеточная лимфома кожи. С перечисленными заболеваниями лимфоматоидный папулез сближает не только общность происхождения, но и нередкое одновременное или последовательное развитие нескольких патологий у одного пациента. Повышенная настороженность дерматологов и онкологов в отношении лимфоматоидного папулеза объясняется возможностью его трансформации в злокачественную Т-клеточную лимфому (в 10-20% случаев).
Причины лимфоматоидного папулеза
Этиология заболевания неизвестна. Патогенез также является предметом споров и обсуждений. Большинство дерматологов, основываясь на гистологической картине лимфоматоидного папулеза, которая выглядит как истинная лимфома, считают заболевание лимфоцитарной опухолью кожи низкой степени злокачественности. Другие рассматривают лимфоматоидный папулез в качестве ограниченной доброкачественной лимфоплазии, возникающей при участии CD4-лимфоцитов, которые регулируют силу иммунного ответа. По всей вероятности, истина находится где-то посередине.
В развитии лимфоматоидного папулеза играет роль нарушение в дифференцировке Т-лимфоцитов, когда в результате латентно текущих аутоиммунных и системных заболеваний, врождённого или приобретённого иммунодефицита, ионизирующего излучения они начинают мутировать, провоцируя усиленную пролиферацию эпидермальных клеток, как здоровых, так и генетически деструктированных. Параллельно изменяются внутренние функции эпидермальных клеток, они начинают продуцировать цитокины и медиаторы, которые запускают механизм воспаления, усиливают пролиферацию клеток поражённой кожи. Так появляются доброкачественные лимфоидные образования дермы.
Экспрессия CD30-антигена атипичными лимфоидными клетками в сочетании с возможностью спонтанного перерождения лимфоматоидного папулеза в злокачественную лимфому свидетельствуют в пользу точки зрения тех дерматологов, которые считают лимфоматоидный папулёз разновидностью опухоли с небольшой степенью злокачественности. С другой стороны, редкость подобной трансформации говорит в пользу теории лимфоплазии. Таким образом, на текущий момент единого мнения не существует. Точно известно лишь то, что если пролиферация лимфоцитов перестанет подчиняться контролю иммунной системы, разовьётся злокачественная лимфома. Это свидетельствует о тесной связи иммунных и онкологических процессов в организме пациента.
Классификация лимфоматоидного папулеза
В современной дерматологии лимфоматоидный папулез классифицируют по гистологической картине, которая помогает в постановке диагноза. Существуют четыре разновидности лимфоматоидного папулеза:
- Тип А – в гистологической картине встречаются разные виды клеток: анапластические лимфоидные клетки, лимфоциты, эозинофилы, нейтрофилы, гистиоциты; процесс смешанноклеточный;
- Тип В – напоминает грибовидный микоз, в гистологическом препарате присутствуют единичные атипичные анапластические клетки злокачественной лимфомы, встречается редко;
- Тип С – гистологически схож с крупноклеточной лимфомой, в препарате преобладают атипичные лимфоидные клетки;
- Тип D – напоминает педжетоидный ретикулёз, наиболее агрессивен из всех типов.
Симптомы лимфоматоидного папулеза
Заболевание дебютирует с высыпания на коже (реже - на слизистых) бессимптомных или слегка зудящих бурых пятен до 3 см в диаметре, на которых в течение нескольких недель появляются узелки, буллы, выпуклые новообразования, покрытые чешуйками. Сыпь не имеет излюбленных мест локализации, появляется на лице, волосистой части кожи головы, конечностях, реже – на слизистых. Элементы сыпи лимфоматоидного папулеза подсыпают волнообразно, способны к саморазрешению и новым высыпаниям, склонны к группированию, особенно часто вокруг волосяных фолликулов.
Полный цикл существования первичных элементов при лимфоматоидном папулезе составляет около 2-8 недель. За это время буллы вскрываются с образованием эрозий, которые покрываются геморрагическими корочками; возможны расчёсы, присоединение вторичной инфекции. Иногда папулы начинают шелушиться - всё это создаёт картину ложного полиморфизма. Каждый элемент сыпи может инволютировать на любой стадии развития. Обычно разрешение первичных элементов начинается с центра, завершается образованием рубца, оставляющего после себя поствоспалительную гипер- или гипопигментацию. Субъективно состояние пациента не нарушается, в редких случаях возможны продромальные явления, зуд.
Диагностика и лечение лимфоматоидного папулеза
Диагностируют лимфоматоидный папулез на основании анамнеза, клиники, данных биопсии кожи, гистологического и иммуногистохимического исследования. Иногда производят биопсию поражённого лимфатического узла, расположенного поблизости. В сомнительных случаях выполняют цитогенетические и молекулярные исследования. Следует отметить, что надёжных критериев, способных предсказать злокачественную трансформацию лимфоматоидного папулеза, сегодня не существует. Дифференцируют заболевание с парапсориазом, лимфомами, крапивницей, медикаментозной токсидермией, чесоткой, синдромом Сезари, лимфогранулематозом.
Радикальных методов терапии лимфоматоидного папулеза нет. Применяют большие дозы тетрациклинов в сочетании с кортикостероидами, сульфонами (дапсон) и цитостатиками (кармустин). Иногда используют ретиноиды, интерфероны. При диссеминированных высыпаниях проводится облучение электронным пучком, ПУВА-терапия. Однако ни один из методов терапии не дает долгосрочного эффекта. Прогноз зависит от течения лимфоматоидного папулеза, количества рецидивов. Необходимо регулярное диспансерное наблюдение у дерматолога.
Впервые лимфоматоидный папулез как самостоятельное заболевание был представлен W. Macaulay в 1968 г. Клиническая картина его характеризуется хроническим длительным течением с периодическими рецидивами высыпаний на коже папулезного и папулонекротического характера, склонных к спонтанному регрессу, после чего почти у всех больных остаются гипо- или гиперпигментированные рубцы. Основной особенностью данного заболевания является относительная доброкачественность клинического течения, несмотря на типичную для злокачественной лимфомы гистологическую картину пролиферата. Изучение данного заболевания с помощью морфологических, иммуноморфологических и электронно-микроскопических исследований позволило установить, что приданной патологии процесс носит Т-лимфопролиферативный характер. В связи с тем, что пролиферирующий злокачественный клон лимфоцитов удается обнаружить лишь у 60% больных, некоторые авторы не рассматривают данное заболевание как истинную ЗЛК, а относят его к псевдолимфомам или пограничным лимфопролиферативным процессам. Однако большинство исследователей в настоящее время полагают, что лимфомато-идный папулез является лимфомой низкой степени злокачественности.
Клинические наблюдения свидетельствуют о том, что лимфоматоидный папулез нередко может быть связан с другими лимфопролиферативными заболеваниями: грибовидным микозом, болезнью Ходжкина, крупноклеточной анапластической лимфомой кожи; при этом может предшествовать указанным заболеваниям, развиваться параллельно с ними или после них.
Гистологически у больных лимфоматоидным папулезом процесс может протекать по трем вариантам. Первый (тип А) — смешанноклеточный — характеризуется наличием клиновидного неэпидермотропного инфильтрата, состоящего из анапластических лимфоидных клеток, малых лимфоцитов, гистиоцитов, эозинофильных и неитрофильных гранулоцитов. Атипичные клетки рассеяны или образуют небольшие скопления. В сосочковом слое дермы наблюдается выраженный отек.
Тип В лимфатоидного папулеза гистологически сходен с грибовидным микозом. Встречается редко, характеризуется полосовидным эпидермотропным инфильтратом, состоящим преимущественно из малых и средних лимфоидных клеток с церебриформными ядрами. Анапластические клетки встречаются редко.
Тип С лимфатоидного папулеза характеризуется пролиферацией крупных лимфоцитов с маркерами CD30+ и морфологически сходен с анапластической крупноклеточной лимфомой кожи В то же время существуют клинические и морфологические различия очагов поражения при указанных заболеваниях. При анапластической крупноклеточной лимфоме количество опухолей незначительно, размеры их более крупные, чем у больных лимфоматоидным папулезом, и обычно превышают 1-1,5 см в диаметре. При морфологическом исследовании кожи, в отличие от истинных СОЗО-положительных крупноклеточных анапластическихлимфом. при которых в дерме обычно наблюдается диффузный инфильтрат, у больных лимфоматоидным папулезом в биоптатах отмечается инфильтрат, располагающийся вокруг сосудов и придатков кожи, со значительным количеством клеток воспалительного характера — нейтрофилов, эозинофилов. Крупные скопления CD30+ клеток у больных лимфоматоидным папулезом обычно отсутствуют.
Крупные анапластические клетки лимфатоидного папулеза имеют Т-клеточное происхождение и кроме маркеров CD30 экспрессируют СD3, СD4 или CD3, CD8.
Злокачественный клон лимфатоидного папулеза генотипически определяется в 65% случаев, специфические хромосомные аномалии не описаны.
Течение и прогноз лимфатоидного папулеза зависят от степени злокачественности пролиферата. Менее благоприятен прогноз при лимфоматоидном папулезе, гистологически сходном с крупноклеточной анагшастической лимфомой кожи. Длительное наблюдение больных лимфоматоидным папулезом (в течение 10-15 лет) свидетельствует о том, что заболевание может трансформироваться в более злокачественные варианты лимфомы: грибовидный микоз, анапластическую крупноклеточную ли мфому кожи или лимфогранулематоз.
Дифференциальный диагноз лимфатоидного папулеза необходимо проводить с каплевидным парапсориазом, папулонекротическим ангиитом и папулонекротическим туберкулезом кожи. Диагноз лимфоматоидного папулеза может быть выяснен при динамическом наблюдении больного, а также с помощью морфологических и иммунофенотипических методов исследования.
Т-клеточная лимфома кожи (ТКЛК) — это разнородная группа заболеваний, относящихся к неходжкинским лимфомам, основной чертой которых является инфильтрация кожи злокачественными моноклональными Т-лимфоцитами.
- Почему развивается Т-клеточная лимфома кожи
- Виды Т-клеточных лимфом кожи и их стадии
- Как проявляется лимфома кожи, симптомы
- Диагностика Т-клеточной лимфомы кожи
- Как проводится лечение лимфомы кожи
- Трансплантация костного мозга и стволовых клеток
- Процесс восстановления после лечения
- Прогноз
Почему развивается Т-клеточная лимфома кожи
Возникновение Т-клеточной лимфомы связано с мутацией зрелых Т-лимфоцитов, что приводит к их бесконтрольному размножению, миграции в кожу и ее инфильтрации. Что именно вызывает мутацию, неизвестно. Предполагается, что она может быть спровоцирована стимуляцией антигенами в результате сбоя работы иммунной системы.
Спровоцировать сбой иммунной системы могут следующие факторы:
- Вирусные инфекции: вирус Эпштейн-Барра, цитомегаловирус, вирус простого герпеса, ретровирусы, лимфотропный вирус человека и др.
- Некоторые химические вещества, которые используются в промышленности, сельском хозяйстве, строительстве и др.
- Ионизирующее излучение, в том числе лучевая терапия в анамнезе.
- Избыточное воздействие УФ-лучей на кожу.
Т-клеточная лимфома кожи может быть первичной и вторичной. Первичные формы начинаются с поражения кожи. При вторичных сначала поражается лимфоидный орган (например, лимфоузлы), в результате чего в нем происходит размножение и накопление лимфоцитов, потом они мигрируют в кожу и инфильтрируют ее.
Виды Т-клеточных лимфом кожи и их стадии
Выделяют следующие виды Т-клеточной лимфомы кожи:
- Грибовидный микоз.
- Синдром Сезари.
- CD30+ Т-клеточные лимфомы — лимфатоидный папулез (ЛП) и кожная анапластическая крупноклеточная лимфома (КАКЛ).
- Панникулитоподобная Т-клеточная лимфома подкожной клетчатки.
- Первичная кожная агрессивная эпидермотропная CD8+ Т-клеточная лимфома.
- Первичная кожная мелко/среднеклеточная CD4+ Т-клеточная лимфома.
Наиболее часто встречаемые первые 2 типа опухоли. При их стадировании заболевания учитываются следующие факторы:
- Степень поражения кожных покровов.
- Вовлеченность в процесс лимфатических узлов.
- Лимфоидное поражение внутренних органов.
- Поражение системы кроветворения.
Их стадирование происходит согласно следующей схеме:
- 1А стадия. Имеются кожные поражения в виде пятен, которые занимают менее 10% кожного покрова, лимфоузлы не увеличены, в крови обнаруживается незначительное количество атипичных клеток Сезари.
- 1В стадия. Помимо пятен на коже, образуются бляшки, но объем высыпаний не превышает 10% от кожного покрова. Остальные признаки такие же, как на 1А стадии.
- 2А стадия. К вышеперечисленным симптомам добавляется поражение лимфатических узлов.
- 2В стадия. На коже появляются узлы.
- 3 стадия. Кожные проявления генерализуются, образуют сливающуюся эритему и покрывают более 90% поверхности тела.
- При 4 стадии, в крови обнаруживается большое количество атипичных клеток Сезари (более 1000 на микролитр), плюс присоединяется лимфоидное поражение внутренних органов.
Пациентов с синдромом Сезари изначально относят к 4 стадии заболевания, поскольку у них имеется обширная эритродермия и большое количество атипичных лимфоцитов в крови.
Остальные нозологии Т-клеточных лимфом, не являющиеся грибовидным микозом и синдромом Сезари, стадируются на основании следующих признаков:
- Т1А — имеется единичный очаг поражения не превышающий 5 см.
- Т1В — единичный очаг поражения, превышающий 5 см.
- Т2 — имеются множественные высыпания кожи, не выходящие за пределы 1-2 зон
- Т2А — зона поражения не превышает 15 см.
- Т2В — зона поражения не превышает 30 см.
- Т2С — высыпания распространяются более чем на 30 см.
- Т3 — имеется поражение кожи, затрагивающее не рядом расположенные зоны, либо высыпания занимают более 3 зон.
Поражение лимфатических узлов:
- N0 — нет увеличения лимфоузлов, ни центральных, ни периферических.
- N1 — поражена 1 группа периферических ЛУ, которые осуществляют лимфодренаж от пораженного участка кожи.
- N2 — поражено более 1 группы ЛУ, липо есть признаки поражения ЛУ, не осуществляющих дренаж вовлеченной области кожи.
- N3 имеются данные за поражение центральных ЛУ.
Поражение внутренних органов:
- М0 — поражения внутренних органов не обнаружено.
- М1 — есть данные за поражение внутренних органов (требуется морфологическое подтверждение).
Как проявляется лимфома кожи, симптомы
Симптоматические проявления будут зависеть от типа Т-клеточной лимфомы.
Симптомы грибовидного микоза:
- Высыпания в виде пятен и бляшек различной формы и цвета. Они носят множественный характер и располагаются на участках кожи, не подвергающихся инсоляции (зона купальника).
- Феномен одновременного прогрессирования и регресса разных элементов сыпи.
- Пойкилодермия — в зоне клеточных высыпаний имеется пятнистая пигментация, расширение кровеносных сосудов и атрофические изменения кожи.
- Наличие зуда.
В зависимости от стадии заболевания, грибовидный микоз будет иметь следующие особенности. На начальном этапе (1 стадия) отмечается наличие множественных или единичных пятен, которые могут достигать размеров в 20 см. Пятна зудят и могут шелушиться. Внешне они напоминают псориаз или экзему. На этой стадии заболевание может существовать годами и даже десятилетиями. Пятна могут спонтанно регрессировать и образовываться снова. Учитывая то, что клиническая картина сходна с другими дерматологическими нозологиями, правильный диагноз сразу поставить сложно, как правило, на это занимает достаточно длительное время.
Для второй стадии пятна начинают трансформироваться в бляшки — пораженная кожа утолщается и как бы приподнимается над здоровой, ее цвет становится красно-синюшным. Бляшки могут спонтанно регрессировать, или, наоборот, увеличиваться в размерах и сливаться друг с другом. На их поверхности может быть шелушение.
На 3 стадии образуются кожные опухоли в виде куполообразных красно-синюшных узлов, с гладкой поверхностью. Узлы увеличиваются в размерах и распадаются, при этом образуются язвы с кровянисто-гнойным отделяемым.
Панникулитоподобная Т-клеточная лимфома подкожной клетчатки проявляется множественными плотными бляшками, либо узлами различного цвета, которые преимущественно располагаются на нижних конечностях. Клинически напоминают панникулит (воспаление подкожной клетчатки).
Первичная CD4+кожная лимфома из мелких/крупных клеток проявляется единичными или множественными узлами синюшно-багрового цвета. В отличие от грибовидного микоза, узлы появляются сразу, без этапа пятна и бляшки.
Клиническим проявлением синдрома Сезари является наличие обширной эритемы (покраснения кожи), которая распространяется не менее чем на 80% покровов тела. Эритема может шелушится, но не обязательно. Характерно увеличение лимфоузлов до размеров ореха. Чаще всего, поражаются паховые и подмышечные ЛУ.
CD30+ Т-клеточная лимфома проявляется рецидивирующими сгруппированными или диссеминированными папулезными высыпаниями. Отдельные элементы могут спонтанно регрессировать в течение нескольких недель, причем на данном фоне могут образовываться новые папулы. В ряде случаев элементы сыпи могут некротизироваться с образованием некротических язв размером от 1 до 4 см, которые самопроизвольно разрешаются с формированием рубца.
Диагностика Т-клеточной лимфомы кожи
Для постановки диагноза необходимо комплексное обследование:
- Осмотр кожных высыпаний.
- Морфологическое и иммунотипическое исследование биоптатов кожи из очагов поражения. Чтобы результаты биопсии были более информативным, необходима отмена всех наружных препаратов, а также системных глюкокортикостероидов за две недели до забора материала. В сомнительных случаях проводят повторную биопсию через 2-4 недели после предыдущей и материал берут из разных очагов.
- Молекулярно-генетические исследования для определения характерных генетических мутаций.
При подозрении на синдром Сезари проводят такое же обследование, как и при грибовидном микозе (ГМ), плюс добавляют следующие данные:
- Определение площади поражения кожи. Если есть узлы, определяют их количество и размер наибольшего из них.
- Производят определение количества клеток Сезари с помощью проточной цитометрии.
Как проводится лечение лимфомы кожи
Для лечения лимфомы кожи используется химиотерапия. Схемы будут определяться типом заболевания и его стадией.
- Топические кортикостероиды. Их необходимо наносить на поверхность пятен и бляшек.
- УФО спектра В — узковолоконное ультрафиолетовое облучение лучами спектра В. Может применяться для пятен и тонких бляшек.
- ПУВА-терапия — псорален + УФО лучами спектра А. Данный вид лечения эффективен при толстых бляшках и фолликулярной форме ГМ.
- Локальная лучевая терапия в СОД 10-20 Гр. Применяется при единичных высыпаниях.
- Тотальная лучевая терапия (ТЛК). Проводится при наличии распространенных высыпаний, которые не реагируют на топические стероиды. Облучение проводится в суммарной очаговой дозе 30-40 Гр с фракционированием разовой очаговой дозы 1,2-1,5Гр. В качестве поддерживающего лечения после ТЛК применяется ПУВА-терапия. Т-клеточная лимфома кожи является чувствительной к облучению опухолью, поэтому лучевая терапия может применяться, как терапия первой линии при лечении ранних и поздних лимфом, так и для лечения рецидивов.
Если наружная терапия лимфомы кожи не оказала эффекта, добавляют вторую линию терапии, в рамках которой могут использоваться следующие системно действующие препараты:
- Ретиноиды.
- Интерфероны.
- Вериностат — ингибитор гистондеацетилазы.
- Цитостатики метотрексат или проспидин.
На поздних стадиях лимфомы кожи используется химиотерапия и электронно-лучевая терапия. Химиотерапия проводится с помощью вориностата. Его необходимо принимать перорально до достижения контроля заболевания (критериями контроля является отсутствие признаков прогрессирования). В случае развития неприемлемых токсических реакций препарат отменяют.
Лечение СС базируется на следующих принципах:
- Комбинированное лечение (наружное и системное) является более эффективным, чем монотерапия.
- По возможности, следует избегать назначения цитостатиков, поскольку они подавляют иммунитет.
- Необходима своевременная диагностика и лечение инфекционных осложнений.
- Поскольку одним из основных симптомов, снижающих качество жизни пациентов, является зуд. Необходимо его эффективное устранение.
Терапия первой линии
Наилучшую эффективность в рамках терапии первой линии у больных с СС показал экстракорпоральный фотоферез (ЭКФ). Из цельной крови выделяют лейкоциты, которые обрабатывают фотосенсибилизирующим веществом и затем обрабатывают светом с заданной длиной волны. Это приводит к их гибели. За одну процедуру можно удалить только часть клеток, поэтому сеансы повторяют с определенной периодичностью — 1 раз в день в течение 2-х дней с последующим 4-х недельным перерывом.
Также может использоваться альфа-интерферон, только в более высоких дозах, чем при лечении ГМ, или метотрексат в низких дозах, при недоступности других видов терапии.
Для достижения лучшего эффекта, эти методы лечения комбинируют с методами наружной терапии, которые используются при ГМ, например:
- Альфа-интерферон + ПУВА.
- Метотрексат + наружные глюкокортикостероиды.
- ЭКФ + ТОК и др.
Терапия второй линии
При отсутствии эффекта от лечения первой линии, переходят ко второй. Здесь уже используются цитостатические препараты:
- Хлорамбуцил.
- Доксорубицин.
- Вориностат.
- Гемцитабин.
- Пентостатин.
- Флударабин+циклофосфамид.
Устранение зуда
Часто больных кожной лимфомой беспокоит зуд. Он может иметь ярко выраженный характер и существенно снижать качество жизни таких людей. Для борьбы с этим симптомом используются следующие препараты и методы терапии:
- Увлажняющие кремы.
- Антигистамины.
- Антибиотики. Доказано, что кожа больных СС обширно колонизирована золотистым стафилококком, поэтому назначение антибактериальных препаратов благоприятно сказывается не только на выраженности зуда, но и на общем течении заболевания.
Если зуд носит нестерпимый характер, назначается габапентин, который используется для лечения боли при нейропатиях. Начинают с дозировки 900 мг/сут, постепенно увеличивая ее до 3600 мг/сут. Для улучшения сна могут назначаться снотворные препараты.
Если имеются многочисленные генерализованные высыпания, используется ПУВА-терапия и низкие дозы метотрексата. Во время терапии наблюдается частичное исчезновение высыпаний, но после отмены лечения, они образуются снова. Полной ремиссии удается достичь редко.
В связи с этим, для контроля лечения необходима поддерживающая терапия с помощью данных методов, но необходимо помнить о возможных осложнениях. Например, ПУВА может спровоцировать развитие рака кожи, а метотрексат — фиброз печени.
Если имеются крупноузелковые элементы (более 2 см), которые не разрешаются самопроизвольно или под действием терапии, их можно удалить хирургически. Тем более при их наличии необходима биопсия для исключения вторичной анапластичной крупноклеточной лимфомы. Альтернативой может стать локальная лучевая терапия. В целом пациенты с ЛП должны пожизненно наблюдаться у врача, поскольку есть вероятность трансформации их заболевания в другие формы лимфом.
При одиночных или сгруппированных высыпаниях показано их хирургическое удаление или локальное облучение. Такая тактика позволяет добиться полной ремиссии у 95% пациентов. Но независимо от применяемого метода лечения, у 40% больных возникают рецидивы. Если они ограничены только кожными проявлениями, без затрагивания лимфатических узлов и внутренних органов, других методов лечения не требуется, можно использовать предыдущую тактику.
Для больных с множественными высыпаниями рекомендованы низкие дозы метотрексата (5-25 мг/нед). Если желаемый эффект не наступает, добавляют терапию альфа-интерфероном. При наличии очагов внекожного проявления необходима системная химиотерапия по протоколу CHOP.
Трансплантация костного мозга и стволовых клеток
При неэффективности других методов лечения, молодым пациентам с поздними стадиями заболевания может быть рекомендована химиотерапия с последующей аллогенной трансплантацией гемопоэтических стволовых клеток (ГСК). Процедура проводится следующим образом.
Первый этап — это химиотерапия, которая призвана уничтожить клон злокачественных Т-лимфоцитов. Этот этап называется кондиционирование. В рамках химиотерапии используются следующие препараты и режимы:
- CHOP — циклофосфамид, доксорубицин, винкристин, преднизолон
- EPOCH — этопозид, циклофосфамид, доксорубицин, винкристин, преднизолон.
- Пентостатин.
- Флурадабин+ интерферон альфа или циклофосфамид.
- Гемцитабин.
Вторым этапом является пересадка гемопоэтических клеток донора. Для реципиента эта процедура не представляет проблем и выглядит как обычное переливание крови. В течение последующих 3-4 недель трансплантат начинает приживаться в костном мозге, о чем свидетельствует повышение уровня лейкоцитов в крови. Генетически, это лейкоциты донора, и они должны полностью заменить иммунную системы больного. Весь этот процесс занимает от нескольких месяцев до года.
Процесс восстановления после лечения
После того как иммунная система восстановится, необходимо будет заново пройти полный курс вакцинации, который выполняется в детском возрасте.
Прогноз
Прогноз заболевания зависит от клинической формы Т-клеточной лимфомы и ее стадии. Для пациентов с ранней стадией ГМ прогноз благоприятный, поскольку эта лимфома кожи очень редко прогрессирует до более серьезных стадий, и средняя продолжительность жизни таких больных составляет 12 лет.
Для пациентов с изначально более поздними стадиями Т-клеточной лимфомы (2В и больше) без признаков поражения внутренних органов, средняя продолжительность жизни составляет 5 лет, при этом для больных с поражением лимфоузлов прогноз несколько хуже, чем при системном обширном поражении кожи. Для больных, у которых лимфома кожи поразила внутренние органы, средняя продолжительность жизни составляет 2,5 года.
CD 30+ Т-клеточные лимфомы характеризуются относительно доброкачественным течением. В частности, лимфоматоидный папулез не влияет на продолжительность жизни больных, за исключением случаев трансформации в другие виды лимфом, в том числе грибовидный микоз, лимфому Ходжкина и др. Трансформация возникает у 4-25% больных и может возникнуть в период дебюта ЛП, после его излечения и даже предшествовать ему. Кожная анапластическая крупноклеточная лимфома также характеризуется благоприятным прогнозом, пятилетней выживаемости достигают 96% больных.
Читайте также: