
Классификация острых лейкозов по fab
Европейская группа иммунологов предложила иммунологическую классификацию острых лейкозов (EGIL, 1995), в основе которой лежит характеристика каждого этапа дифференцировки клеток-предшественниц гемопоэза по наличию на их мембране определенного набора антигенов дифференцировки – CD (cluster of differentation – кластер дифференцировки). К антигенам, выявляющимся на клетках лимфоидной линии относятся CD1 – CD5, CD7 – CD10, CD20, CD22, CD23, CD53, CD57, миелоидной – CD11, CD13 – CD15, CD33, CD36, CD41, CD42, CD65, HLA-DR, стволово-клеточный антигенный маркер CD34.
В развитии острых лейкозов различают следующие стадии: начальную, развернутую, полную ремиссию, частичную ремиссию, рецидив, терминальную стадию, выздоровление (состояние полной ремиссии на протяжении 5 лет и более). При хронических лейкозах частично задерживается созревание клеток, субстрат опухоли составляют созревающие и зрелые клетки, которые в основном и обнаруживаются в периферической крови, анемия в большинстве случаев развивается по мере прогрессирования заболевания. Более медленное течение хронических лейкозов прогностически не оказывается более благоприятно. Во многих случаях острые лейкозы успешно лечатся, в то время как хронические оказываются резистентными к терапии.
Острые и хронические лейкозы развиваются на разной клональной и неидентичной мутационной основе. Острый лейкоз с течением времени не переходит в хронический, поскольку утраченную раннее способность к дифференцировке неоплазма вновь не приобретает. Хронический лейкоз также никогда не обостряется.
По количеству лейкоцитов в периферической крови (В. Демешек, цитируется по А.Ш. Зайчику и А.П. Чурилову, 2002) лейкозы на той или иной стадии их течения квалифицируют как:
· лейкемические (резкое увеличение количества лейкоцитов – 100,0 x 10 9 /л и выше);
· сублейкемические (увеличение числа лейкоцитов до 100,0 x 10 9 /л);
· алейкемические (число лейкоцитов не изменено);
· лейкопенические (число лейкоцитов уменьшено – 9 /л).
Острый лимфобластный лейкоз (ОЛЛ).
ОЛЛ– опухоль, развивающаяся из клетки-предшественницы лимфопоэза. У взрослых она бывает редко; в детском возрасте составляет 80% всех форм лейкозов. Пик заболеваемости приходится на возраст 4-5 лет. Заболевают чаще дети с иммунологической недостаточностью, с хромосомными аномалиями.
Различают (Fab, 1976) 3 морфологических варианта ОЛЛ – L1, L2 , L3, дифференциальным признаком которых является размер, форма ядер, структура ядерного хроматина, степень выраженности нуклеол, ядерно-плазматические соотношения, вакуолизация цитоплазмы лейкемических клеток.
В соответствии с иммунологической классификацией различают по 4 варианта Т-ОЛЛ и В- ОЛЛ.
Лейкозные лимфобласты вытесняют миелоидные элементы из костного мозга и замещают их в периферической крови. Обнаруживаются бластные клетки с аномальным кариотипом (анэуплоидия, изменение структуры хромосом). Наиболее важной аномалией является транслокация t(9;22) – филадельфийская хромосома, первоначально описанная как характерная для хронического миелолейкоза. Бласты с такой аномалией встречаются у 5% больных ОЛЛ детей и 30% взрослых. Для пациентов, бласты которых несут подобную транслокацию прогноз неблагоприятный.
В картине крови при ОЛЛ отмечаются анемия, тромбоцитопения, гранулоцитопения, лимфобласты L1 – L3 морфологических типов; значительно снижено содержание дифференцированных лимфоцитов – абсолютная лимфопения. При низком уровне лейкоцитов не всегда выявляются бласты. В этом случае только исследование костного мозга может подтвердить диагноз ОЛЛ (много лимфобластов, отсутствуют или резко снижены миелоидные элементы). Кроме того в клинической картине присутствуют гипоксический, геморрагический, инфекционно-септический синдромы, паранеопластические симптомы, обусловленные цитокинами, выделяемыми клетками иммунной системы, лейкозными бластами (анорексия, исхудание, остеопороз, костные боли). Обнаруживаются также лимфаденопатия, гепатоспленомегалия, менингеальные явления, нейролейкемия.
Прогноз. Наиболее прогностически благоприятна форма L1, менее – L3; при L2 прогноз промежуточный, вероятность стойкой ремиссии 50%.
Острый миелоидный лейкоз.
Острый миелоидный лейкоз (также ОМЛ, острый нелимфобластный лейкоз, острый миелогенный лейкоз) — это злокачественная опухоль миелоидного ростка крови, при которой быстро размножаются изменённые белые кровяные клетки. Накапливаясь в костном мозге, они подавляют рост нормальных клеток крови, что приводит к снижению количества эритроцитов, тромбоцитов, и нормальных лейкоцитов. Болезнь проявляется быстрой утомляемостью, одышкой, частыми мелкими повреждениями кожи, повышенной кровоточивостью, частыми инфекционными поражениями. До сих пор явная причина заболевания неизвестна, однако некоторые факторы риска его возникновения выявлены. ОМЛ является острым заболеванием, развивается быстро, и без лечения приводит к смерти больного за несколько месяцев, иногда — недель.
Это самый распространённый вид острого лейкоза у взрослых, заболеваемость им с возрастом увеличивается.
Встречаются несколько разновидностей ОМЛ, лечение и прогноз для них оказывается разным.
Франко-американско-британская классификационная (Fab) система разделяет ОМЛ на 9 подвидов, от М0 по M8, основываясь на типах клеток — предшественниц лейкоцитов, и на степени зрелости изменённых клеток. Определение злокачественных клеток проводят на основании внешних признаков при световой микроскопии и/или цитогенетически, выявляя лежащие в основе отклонений изменения в хромосомах. У разных подвидов ОМЛ разные прогноз и ответ на лечение.
Подвиды острого миелоидного лейкоза по классификации ВОЗ:
· ОМЛ с характернымигенетическими изменениями
· ОМЛ с изменениями, связанными смиелодисплазией
· ОМЛ и миелодиспластический синдром, связанные спредыдущим лечением
· Миелоидная саркома (хлорома)
· Миелопролиферативные заболевания, связанные ссиндромом Дауна
· Бластная плазмацитоидная дендритноклеточная опухоль
· ОМЛ,не подпадающие под признаки перечисленных подвидов
o Острый миелобластный лейкоз с минимальной дифференциацией
o Острый миелобластный лейкоз без созревания
o Острый миелобластный лейкоз с созреванием
o Острый миеломоноцитарный лейкоз
o Острый монобластный лейкоз и острый моноцитарный лейкоз
o Острый эритроидный лейкоз
o Острый мегакариобластный лейкоз
o Острый базофильный лейкоз
o Острый эозинофильный лейкоз
o Острый панмиелоз с миелофиброзом
Бывают такие подвиды острого лейкоза, при которых изменённые лейкоциты невозможно определить как лимфоциты или гранулоциты, или когда присутствуют злокачественно изменённые клетки обоих ростков. Такие лейкозы иногда называют бифенотипными острыми лейкозами.
Последнее изменение этой страницы: 2016-12-30; Нарушение авторского права страницы
В 1976 г. группой экспертов Франции, США и Великобритании (FAB) была предложена классификация острых лейкозов, основанная на морфологических и цитохимических особенностях бластных клеток.
В настоящее время следует использовать также результаты иммунофенотипирования и цитогенетического исследования, необходимые для определения индивидуального прогноза и выбора адекватной терапии (интенсификация, миелот-рансплантация или стандартное лечение, которое часто включает длительную поддерживающую химиотерапию).
В соответствии с FAB-классификацией на основании морфологического исследования крови и костного мозга и цитохимических реакций выделяют 8 типов острого миелобластного лейкоза (ОМЛ) (М0-М7) и 3 типа острого лимфобластного лейкоза (L1-L3). В настоящее время FAB-классификация ОЛЛ не используется; при ОМЛ практически всегда дополнительно используются иммунофенотипирование и цитогенетическое исследование бластных клеток.
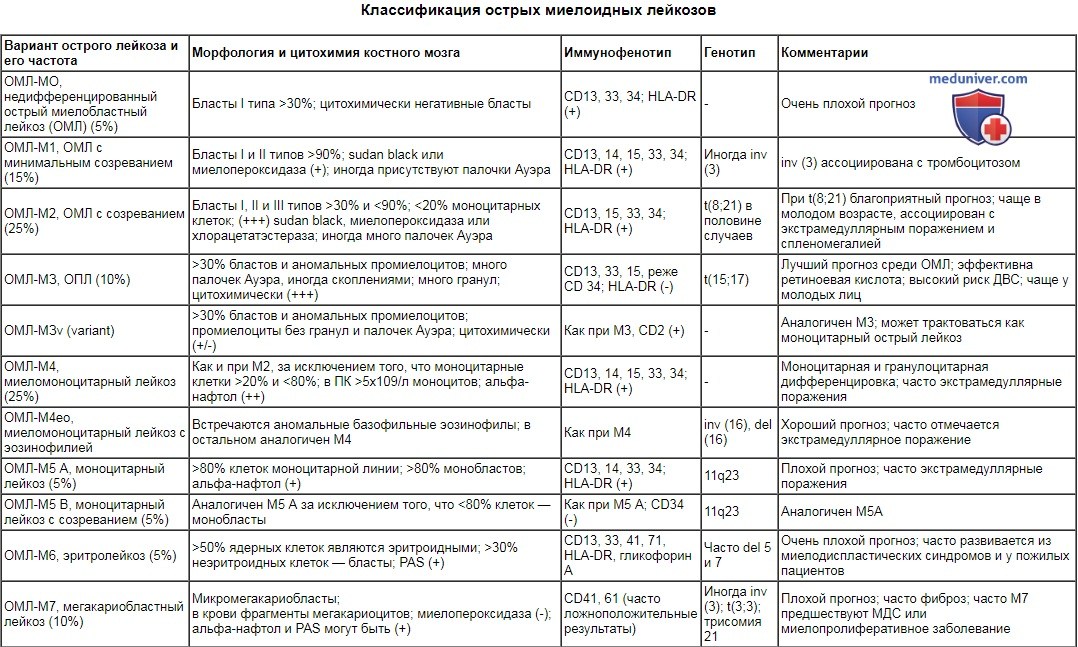
| Вариант острого лейкоза и его частота | Морфология и цитохимия костного мозга | Иммунофенотип | Генотип | Комментарии |
| ОМЛ-МО, недифференцированный острый миелобластный лейкоз (ОМЛ) (5%) | Бласты I типа >30%; цитохимически негативные бласты | CD13, 33, 34; HLA-DR (+) | - | Очень плохой прогноз |
| ОМЛ-М1, ОМЛ с минимальным созреванием (15%) | Бласты I и II типов >90%; sudan black или миелопероксидаза (+); иногда присутствуют палочки Ауэра | CD13, 14, 15, 33, 34; HLA-DR (+) | Иногда inv (3) | inv (3) ассоциирована с тромбоцитозом |
| ОМЛ-М2, ОМЛ с созреванием (25%) | Бласты I, II и III типов >30% и 30% бластов и аномальных промиелоцитов; много палочек Ауэра, иногда скоплениями; много гранул; цитохимически (+++) | CD13, 33, 15, реже CD 34; HLA-DR (-) | t(15;17) | Лучший прогноз среди ОМЛ; эффективна ретиноевая кислота; высокий риск ДВС; чаще у молодых лиц |
| ОМЛ-МЗv (variant) | >30% бластов и аномальных промиелоцитов; промиелоциты без гранул и палочек Ауэра; цитохимически (+/-) | Как при М3, CD2 (+) | - | Аналогичен М3; может трактоваться как моноцитарный острый лейкоз |
| ОМЛ-М4, миеломоноцитарный лейкоз (25%) | Как и при М2, за исключением того, что моноцитарные клетки >20% и 5х109/л моноцитов; альфа-нафтол (++) | CD13, 14, 15, 33, 34; HLA-DR (+) | - | Моноцитарная и гранулоцитарная дифференцировка; часто экстрамедуллярные поражения |
| ОМЛ-М4ео, миеломоноцитарный лейкоз с эозинофилией | Встречаются аномальные базофильные эозинофилы; в остальном аналогичен М4 | Как при М4 | inv (16), del (16) | Хороший прогноз; часто отмечается экстрамедуллярное поражение |
| ОМЛ-М5 А, моноцитарный лейкоз (5%) | >80% клеток моноцитарной линии; >80% монобластов; альфа-нафтол (+) | CD13, 14, 33, 34; HLA-DR (+) | 11q23 | Плохой прогноз; часто экстрамедуллярные поражения |
| ОМЛ-М5 В, моноцитарный лейкоз с созреванием (5%) | Аналогичен М5 А за исключением того, что 50% ядерных клеток являются эритроидными; >30% неэритроидных клеток — бласты; PAS (+) | CD13, 33, 41, 71, HLA-DR, гликофорин А | Часто del 5 и 7 | Очень плохой прогноз; часто развивается из миелодиспластических синдромов и у пожилых пациентов |
| ОМЛ-М7, мегакариобластный лейкоз (10%) | Микромегакариобласты; в крови фрагменты мегакариоцитов; миелопероксидаза (-); альфа-нафтол и PAS могут быть (+) | CD41, 61 (часто ложноположительные результаты) | Иногда inv (3); t(3;3); трисомия 21 | Плохой прогноз; часто фиброз; часто М7 предшествуют МДС или миелопролиферативное заболевание |
Наиболее важным для дифференциальной диагностики является выделение лимфоидной или миелоидной принадлежности острого лейкоза, так как имеются значительные различия в лечении больных ОМЛ и ОЛЛ. В связи с тем что современные программы терапии для всех морфологических типов ОМЛ, за исключением ОПЛ, обычно схожи, а прогноз, как правило, неблагоприятный, использование лишь FAB-классификации нецелесообразно.

Необходимо также иметь в виду, что у части пациентов морфологические и цитохимические исследования, проведенные квалифицированными врачами-лаборантами, либо не позволяют верифицировать вариант ОЛ, либо дают ошибочный диагноз у части больных (при изучении препаратов периферической крови и костного мозга различными экспертами диагноз подтверждается не более чем в 80-90% случаев).
В этих случаях показано иммунофенотипирование бластных клеток. Наконец, в настоящее время программы цитостатической терапии дифференцированы в зависимости от иммунологического варианта (прежде всего при ОЛЛ) и характера цитогенетических изменений. По всем этим причинам морфологическое и цитохимическое исследование крови и костного мозга в большинстве случаев должно дополняться иммунофенотипированием и цитогенетическими исследованиями.
*Импакт фактор за 2018 г. по данным РИНЦ
Журнал входит в Перечень рецензируемых научных изданий ВАК.
Читайте в новом номере
Острый лейкоз - заболевание, в основе которого лежит образование клона злокачественных (бластных) клеток, имеющих общую клетку-предшественницу. Бласты инфильтрируют костный мозг, вытесняя постепенно нормальные гемопоэтические клетки, что приводит к резкому угнетению кроветворения. Для многих типов лейкозов характерна также бластная инфильтрация внутренних органов.
Острый лейкоз подразделяют на лимфобластный (ОЛЛ) и миелобластный (ОМЛ). Считается, что возникновение острого лейкоза могут обусловить следующие факторы:
- неустановленные (чаще всего);
- наследственные:
- синдром Дауна
- синдром Блума
- анемия Фанкони
- атаксия-телеангиэктазия
- синдром Клайнфелтера
- несовершенный остеогенез
- синдром Вискотта - Олдрича
- лейкоз у близнецов
- химические:
- бензол
- алкилирующие агенты (хлорамбуцил, мельфалан)
- радиоактивное облучение
- предрасполагающие гематологические расстройства (миелодисплазия, апластическая анемия)
- вирусы HTLV-I, вызывающие Т-клеточный лейкоз и лимфому у взрослых.
В последние десятилетия достигнуты значительные успехи в лечении острых лейкозов. Пятилетняя выживаемость зависит от типа лейкоза и возраста пациентов:
- ОЛЛ у детей - 65 - 75%;
- ОЛЛ у взрослых - 20 - 35%;
- ОМЛ у пациентов моложе 55 лет - 40 - 60%;
- ОМЛ у пациентов старше 55 лет - 20%.
Классификация
Различия между ОЛЛ и ОМЛ базируются на морфологических, цитохимических и иммунологических особенностях названных типов лейкозов. Точное определение типа лейкоза имеет первостепенное значение для терапии и прогноза.
Как ОЛЛ, так и ОМЛ в свою очередь подразделяются на несколько вариантов согласно FAB- классификации (French-American-British). Так, существуют три варианта ОЛЛ - L1, L2, L3 и семь вариантов ОМЛ:
- М0 - недифференцированный ОМЛ;
- М1 - миелобластный лейкоз без созревания клеток;
- М2 - миелобластный лейкоз с неполным созреванием клеток;
- М3 - промиелоцитарный лейкоз;
- М4 - миеломоноцитарный лейкоз;
- М5 - монобластный лейкоз;
- М6 - эритролейкоз;
- М7 - мегакариобластный лейкоз.
В соответствии с экспрессируемыми антигенами ОЛЛ делится на Т-клеточный и В-клеточный типы, включающие в себя в зависимости от степени зрелости несколько подтипов (пре-Т-клеточный, Т-клеточный, ранний пре-В-клеточный, пре-В-клеточный, В-клеточный). Четкая корреляция между морфологическими и иммунофенотипическими вариантами отсутствует, за исключением того, что морфология L3 характерна для В-клеточного лейкоза.
Что касается ОМЛ, иммунофенотипирование (т.е. определение экспрессируемых антигенов) не всегда помогает различить варианты М0 - М5. С этой целью дополнительно используют специальное цитохимическое окрашивание. Для постановки диагноза эритролейкоза (М6) и мегакариобластного лейкоза (М7) бывает достаточно иммунофенотипирования.
Распространенность
ОЛЛ наиболее часто возникает в возрасте 2 - 10 лет (пик в 3 - 4 года), затем распространенность заболевания снижается, однако после 40 лет отмечается повторный подъем. ОЛЛ составляет около 85% лейкозов, встречающихся у детей. ОМЛ, напротив, наиболее часто встречается у взрослых, причем частота его увеличивается с возрастом.
Клинические проявления
Клинические проявления при лейкозах обусловлены бластной инфильтрацией костного мозга и внутренних органов. Анемия проявляется бледностью, вялостью, одышкой. Нейтропения приводит к различным инфекционным осложнениям. Основные проявления тромбоцитопении - спонтанное образование гематом, кровотечения из носа, матки, мест инъекций, десен. Характерны также боли в костях, лимфаденопатия, гепатоспленомегалия. Возможны затруднение дыхания в связи с наличием медиастинальных масс, увеличение яичек, менингеальные симптомы. При ОМЛ встречается гипертрофия десен.
Обследование пациентов
Общий анализ крови: возможно снижение уровня гемоглобина и числа тромбоцитов; содержание лейкоцитов - от менее 1,0 • 10 9 /л до 200 • 10 9 /л, дифференцировка их нарушена, присутствуют бласты.
Коагулограмма может быть изменена, особенно при промиелоцитарном лейкозе, когда в бластных клетках имеются гранулы, содержащие прокоагулянты.
Биохимический анализ крови при высоком лейкоцитозе может свидетельствовать о почечной недостаточности.
Рентгенограмма органов грудной клетки позволяет выявить медиастинальные массы, которые встречаются у 70% больных с Т-клеточным лейкозом.
Костномозговая пункция: гиперклеточность с преобладанием бластов.
Иммунофенотипирование - определяющий метод в разграничении ОЛЛ и ОМЛ.
Цитогенетические и молекулярные исследования позволяют выявлять хромосомные аномалии, например филадельфийскую хромосому (продукт транслокации части 9-й хромосомы на 22-ю; определяет плохой прогноз при ОЛЛ).
Люмбальная пункция используется для выявления поражения центральной нервной системы (нейролейкоз).
Все пациенты с подозреваемым или установленным лейкозом должны быть как можно быстрее направлены для обследования и лечения в специализированные стационары.
Поддерживающая терапия предусматривает трансфузии тромбоцитов, эритроцитов, свежезамороженной плазмы, антибиотикотерапию инфекционных осложнений.
| Факторы | ОЛЛ | ОМЛ |
| Возраст | Менее 1 года или более10 лет | Более 60 лет |
| Пол | Мужской | Мужской или женский |
| Лейкоцитоз | Более 50 • 10 9 /л | Более 50 • 10 9 /л |
| Поражение ЦНС | Бласты в ликворе | Бласты в ликворе |
| Ремиссия | Не достигнута после индукционной терапии | Более 20% бластов в костном мозге после 1-го курса лечения |
| Цитогенетика | Филадельфийская хромосома | Делеции или моносомия по 5 или 7 хромосомам; множественные хромосомные аномалии |
Цель химиотерапии - индукция ремиссии (менее 5% бластов в костном мозге) и последующая элиминация резидуальных бластных клеток посредством консолидирующей терапии. Химиопрепараты нарушают способность злокачественных клеток к делению, а комбинирование двух или трех препаратов повышает эффективность терапии и снижает риск развития резистентности бластов к терапии. Для профилактики и лечения нейролейкоза используются эндолюмбальные введения метотрексата и краниальное облучение.
Трансплантация костного мозга (ТКМ). Аллогенная ТКМ может применяться в случае плохого прогноза при ОЛЛ, при ОМЛ в первую ремиссию, при рецидивах лейкозов. Однако в связи с дефицитом совместимых доноров эта возможность доступна далеко не всем пациентам.
Факторы,обусловливающие плохой прогноз при острых лейкозах, представлены в таблице.
Токсичность терапии
Ранняя токсичность включает тошноту, рвоту, мукозиты, выпадение волос, нейропатии, печеночную и почечную недостаточность, выраженное угнетение кроветворения.
Поздняя токсичность может проявляться поражениями различных органов:
- сердце - аритмии, кардиомиопатии;
- легкие - фиброз;
- эндокринная система - задержка роста, гипотиреоидизм, бесплодие;
- почки - снижение гломерулярной фильтрации;
- психика - эмоциональные и интеллектуальные нарушения;
- вторичные опухоли;
- катаракта.
Все пациенты с острым лейкозом должны наблюдаться не менее 10 лет по завершении лечения. Особого внимания требуют к себе такие проблемы, как задержка роста и эндокринные дисфункции у детей.
Liesner RJ, Goldstone AH. The acute leukaemias. BMJ 1997;314:733-6.
Для того чтобы практический врач мог ориентироваться в новой классификации острых лейкозов, предложенной ВОЗ, приводим старую ФАБ-классификацию, которая предусматривала выделение следующих форм ОнЛЛ.
■ М0 — острый миелобластный лейкоз с минимальной миелоидной диф-ференцировкой. При данной форме лейкоза бласты без зернистости составляют более 30% миелокариоцитов. Менее 3% бластов содержат липиды или миелопероксидазу. Бласты относятся к миелобластам по результатам фенотипирования (CD13+, CD33+).
■ М1 — острый миелобластный лейкоз без созревания. Бласты без зернистости или с единичными азурофильными гранулами, могут содержать тельца Ауэра; нуклеолы единичные. Бласты должны составлять 90% или более из неэритропоэтических клеток. Более 3% бластов перокси-дазоположительны и содержат липиды.
■ М2 — острый миелобластный лейкоз с созреванием. Бласты морфологически и цитохимически не отличаются от М1, составляют от 30 до 89% неэритропоэтических клеток. Палочки Ауэра, как правило, единичные, обычные. Миелоциты, метамиелоциты и гранулоциты могут быть выявлены в вариабельном количестве (более 10%) и часто имеют ненормальную морфологию. Моноцитарные клетки составляют менее 20% неэритропоэтических клеток.
■ М3 — острый промиелоцитарный лейкоз. Большая часть клеток соответствует неопластическим промиелоцитам. Клетки часто разрушены, так что можно выявить свободно расположенные гранулы и палочки Ауэра. Ядра бластов расположены эксцентрично, варьируют в форме и размере, часто состоят из двух долей.
■ М4 — острый миеломонобластный лейкоз. Общее количество бластов в костном мозге составляет более 30%, при этом более 20% бластов костного мозга и/или более 5х10 9 /л клеток периферической крови — монобласты, промоноциты или моноциты. Диагноз М4 ставят в том случае, когда изменения в костном мозге соответствуют М2, но в периферической крови обнаруживают более 5,0х10 9 /л моноцитарных клеток. Промоноциты и моноциты отличаются отчётливой диффузной реакцией на наличие а-нафтилацетатэстеразы, ингибируемой NaF. Характерный признак М4 — увеличение концентрации лизоцима в крови и моче более чем в 3 раза.
■ М5 — острый монобластный лейкоз. Бласты составляют более 30% ми-елокариоцитов. В костном мозге 80% и более неэритроидных клеток составляют монобласты, промоноциты и моноциты. М5 по типу блас-тов разделяют на две формы: М5а — монобласты составляют 80% или более всех бластов; М5б — монобласты составляют менее 80%, а остальные — промоноциты и моноциты, причём последние составляют в среднем 20% бластов.
■ Мб — острый эритромиелоз. В красном костном мозге эритрокариоци-ты составляют более 50% всех клеток, характеризуются дольчатостью и фрагментацией ядра, многоядерностью, гигантскими формами. Бласты составляют более 30% неэритроидных клеток и могут относиться к любому из ФАБ-вариантов бластов, кроме М3. Такие эритробласты часто выходят в периферическую кровь. Для эритрокариоцитов характерна диффузно-гранулярная реакция на а-нафтилацетатэстеразу.
■ М7 — острый мегакариобластный лейкоз (введён в ФАБ-классификацию в 1985 г.). Свыше 30% клеток составляют незрелые, очень полиморфные бласты. Часто сильно базофильная цитоплазма бластов образует псевдоподии. Рутинная цитохимия не показательна. Часто выявляют миелофиброз.
В табл. приведены основные классификации острых лейкозов.
Таблица Классификации острых лейкозов
Таблица Классификации острых лейкозов
А.И. Воробьев, М.Д. Бриллиант (2000)
Острые миелоидные лейкозы
Острый миеломонобласт-ный лейкоз, вариант с t(8;21) (q22;q22) и вариант с перестройками 11q23
ОМЛ с t(8;21) (q22;q22) ОМЛ с перестройками 11q23 ОМЛ:
с мультилинейной дисплазией; с предшествующим миелодис-пластическим синдромом; без предшествующего миелодис-пластического синдрома ОМЛ с минимальной дифференцировкой ОМЛ без признаков вызревания
Острый промиелоцитарный лейкоз с t(15;17)(q22;q11-12) и вариантами
ОМЛ с признаками вызревания ОМЛ с базофилией Промиелоцитарный лейкоз (ОМЛ) с t(15;17)(q22;q11-12) и вариантами
Острый миеломонобластный лейкоз
вариант с inv(16)(p13;q22) или t(16;16)p13;q22) и с патологической костномозговой эозинофилией; вариант с перестройками 11q23
ОМЛ с inv(16)(p13;q22) или t(16;16)p13;q22) и с патологической костномозговой эозинофилией
ОМЛ с перестройками 11q23
Острый монобластный лейкоз, вариант с перестройками 11q23
Острый моноцитарный лейкоз ОМЛ с перестройками 11q23
Острый эритромиелоз Острый эритромегакариобласт-ный лейкоз
Острый эритроидный лейкоз
Острый монобластный лейкоз
Острый мегакариоцитарный лейкоз
Острый мегакариобластный лейкоз с миелофиброзом
Острый мегакариоцитарный лейкоз
Острый миелобластный лейкоз
Острый панмиелоз с миелофибро-
рефрактерная анемия с кольцевыми сидеробластами без кольцевых сидеробластов рефрактерная цитопения (миело-диспластический синдром) с мультилинейной дисплазией рефрактерная анемия (миелодис-пластический синдром) с избытком бластов синдром 5q-
миелодиспластические синдромы неквалифицируемые
Общие сведения о острых лейкозах
Острые лейкозы (ОЛ) - опухолевые клональные заболевания гемопоэтической ткани, при котором лейкемическая трансформация генетического аппарата происходит на уровне мультилинейной стволовой или коммитированной клетки-предшественницы.
Для них характерно первичное поражение костного мозга (КМ) морфологически незрелыми кроветворными (бластными) клетками с вытеснением ими нормальных элементов и инфильтрацией различных органов и тканей.
Принадлежность бластных клеток к той или иной линии кроветворения, степень их дифференцировки обуславливают клиническое течение острого лейкоза, проводимую терапию, эффективность лечения и прогноз.
Диагностика острых лейкозов включает исследование периферической крови, КМ и в отдельных случаях - проведение трепанобиопсии. Анемия, нейтропения и тромбоцитопения обычно присутствуют у большинства больных ОЛ. Морфологическая оценка состава пунктата костного мозга является базовой при диагностике острых лейкозов.
Без подсчета миелограммы нельзя интерпретировать данные других методов исследования. Характерным признаком ОЛ является бластная метаплазия. Для установления диагноза острый лейкоз число бластов в миелограмме должно превышать 20%, независимо от их наличия или отсутствия в периферической крови.
Современный алгоритм диагностики и дифференциальной диагностики вариантов острых лейкозов включает следующие методы исследования:
- морфологический,
- цитохимический,
- иммунофенотипический,
- цитогенетический.
К морфологическим критериям характеристики бластов относятся:
- размер клеток (макро, мезо, микрогенерации),
- форма ядер (округлая, складчатая, моноцитоидная),
- наличие зернистости и/или палочек Ауэра в цитоплазме,
- ядерно-цитоплазматическое соотношение (высокое, умеренное, низкое).
Именно на основании морфологических признаков в случаях уточнения вариантов острого миелоидного лейкоза (ОМЛ) лейкемические миелобласты и монобласты разделяются на клетки с наличием или отсутствием признаков созревания.
Основой современной диагностики острых лейкозов послужила FAB-классификация, предложенная в 1976 г. После ее пересмотра в 1991 г. установлены критерии выделения 8 типов - М0-М7-острых миелоидных и трех типов Л1-Л3, В- и Т-острых лимфобластных лейкозов (ОЛЛ).
Классификация
Классификация ОЛ (1997г.) базируется на данных морфологического, цитохимического, иммунологического и цитогенетического исследований.
Общепринятым является выделение следующих вариантов ОЛ:
М0 - миелобластный с минимальной дифференцировкой,
М1 - миелобластный без созревания,
М2 - миелобластный с созреванием,
М2 баз. - базофильно-клеточный,
М3 - промиелоцитарный,
М4 - миеломонобластный,
М5а - монобластный без созревания,
М5в - монобластный с созреванием,
М6 - эритромиелоз,
М7 - мегакариобластный,
Л1 - В- и Т-линейный,
Л2 - В- и Т-линейный,
Л3 - В-линейный типа лимфомы Беркитта.
В 2008 году Всемирная организация здравоохранения (ВОЗ) приняла новую систему классификации миелоидных и лимфоидных новообразований. Основные принципы этой системы состояли в том, чтобы при классификации использовать не только морфологические характеристики, но и дополнительную информацию, включая клинические, генетические, иммунофенотипические и молекулярно-биологические данные для определения специфических нозологических форм заболеваний.
Подобно классификации FAB, система ВОЗ основывается на морфологических, цитохимических и иммунофенотипических характеристиках неопластических клеток, принадлежащих к определенной клеточной линии, находящейся на определенной стадии дифференцировки.
Однако имеются и некоторые отличия. Определение процента бластов с учетом степени их зрелости и диспластических аномалий рекомендуется проводить путем дифференцированного подсчета 200 нейтрофильных лейкоцитов периферической крови и 500 клеток миелоидного ростка в препаратах костного мозга.
В число бластов периферической крови и КМ не включены клетки, экспрессирующие антиген CD34; хотя все гемопоэтические клетки CD34+ являются бластами, но не все бласты экспрессируют антиген CD34.
Цитохимическая диагностика (миелопероксидаза, неспецифические эстеразы и др.) и/или иммунофенотипирование (определение миелоидных антигенов, таких как CD13, CD33, CD 117 и др.) могут иметь значение в определении принадлежности бластных клеток к одной или нескольким миелоидным линиям, что дополняет специфические морфологические данные (например, наличие палочек Ауэра).
Хотя FAB-классификация дает возможность распознавания морфологической гетерогенности ОМЛ, она не отражает генетической или клинической разнородности заболевания.
Некоторые исследователи считают, что в ОМЛ целесообразно выделение двух подгрупп в зависимости от наличия или отсутствия миелодиспластического синдрома:
- острый миелоидный лейкоз, которому предшествует МДС или который имеет признаки МДС,
- ОМЛ, который возникает de novo без признаков миелодисплазии.
Характеристики, связанные с этими двумя подгруппами, показывают, что имеются два фундаментально различных механизма лейкомогенеза. МДС-обусловленный ОМЛ связан с мультилинейной дисплазией, цитогенетическими данными за неблагоприятный прогноз, что часто включает потерю цитогенетического материала, и плохим ответом на проводимую терапию.
При ОМЛ de novo мультилинейная дисплазия отсутствует, часто имеется наличие генетических аномалий группы благоприятного прогноза, в частности - наличие инверсий и обратимых хромосомных транслокаций, с хорошим ответом на терапию, с длительной безрецидивной и общей выживаемостью.
Этот тип острого миелоидного лейкоза стабильно проявляется в любой возрастной группе, но чаще встречается в детском и молодом возрасте. Возможно, что специфические геномные изменения, ассоциированные с этим вариантом лейкоза, с частым вовлечением в процесс факторов транскрипции, играют большую роль в его патогенезе.
Показано, что пациенты с количеством бластов от 20 до 29% в крови или КМ часто имеют сходные клинические данные, а также сходный ответ на терапию и имеют одинаковую продолжительность жизни в сравнении с пациентами, которые имеют 30% или более бластов.
Согласно FAB-классификации, пациентов с количеством бластов 20-29% относят к группе рефрактерной анемии с избытком бластов в стадии бласттрансформации (РАИБТ). По классификации ВОЗ, у большинства пациентов с 20-29% бластов заболевание классифицируют как ОМЛ с мультилинейной дисплазией - подгруппа включает пациентов как с предшествующим МДС, так и пациентов с первичным ОМЛ и дисплазией множественных клеточных линий. ОМЛ с мультилинейной дисплазией рассматривается как более выраженное проявление МДС.
Миелоидные клетки при РАИБТ и ОМЛ с предшествующим МДС имеют идентичные профили пролиферации и апоптоза, что отличает их от клеток при рефрактерной анемии (РА), рефрактерной анемии с кольцевыми сидеробластами (РАКС) и рефрактерной анемии с избытком бластов (РАИБ). При этом часто повышена экспрессия протеина множественной лекарственной устойчивости (МЛУ).
Основные различия между классификацией ВОЗ и FAB-классификацией:
- снижение процента бластов в периферической крови или костном мозге с 30 до 20%,
- распределение ОМЛ по клиническим и биологическим подгруппам,
- при наличии клональных обратимых цитогенетических аномалий: t (8; 21) (q22; q22), inv (16) (p13; q22), t (16; 16) (p13; q22), t (15; 17) (q22; q12) диагноз ОМЛ выставляется независимо от процента бластов в периферической крови и/или костном мозге;
- пациенты с 20-29% бластов и мультилинейной дисплазией отнесены к группе ОМЛ с мультилинейной дисплазией - эта подгруппа включает как пациентов с предшествующим МДС, так и пациентов с первичным ОМЛ и дисплазией нескольких клеточных линий.
Острые миелоидные лейкозы, опухоли из клеток-предшественников и острые лейкозы неопределенных клеточных линий (классификация ВОЗ, 2008 г.):
• острый миелоидный лейкоз с повторяющимися генетическими аномалиями:
- острый миелоидный лейкоз с t (8; 21) (q22; q22); RUNX1-RUNX1T1,
- острый миелоидный лейкоз с inv (16) (p13,1; q22) or t (16; 16) (p13,1; q22); CBF-MYH11,
- острый промиелоцитарный лейкоз с t (15; 17) (q22; q12); PML-RARA,
- острый миелоидный лейкоз с t (9; 11) (p22; q23); MLLT3-MLL,
- острый миелоидный лейкоз с t (6; 9) (p23; q34); DEK-NUP214,
- острый миелоидный лейкоз с inv (3) (q21; q26,2) или t (3; 3) (q21; q26,2); RPN1-EVI1,
- острый (мегакариобластный) лейкоз с t (1; 22) (p13; q13); RBMT15-MKL1,
- условная подгруппа: острый миелоидный лейкоз с мутантной NPM1,
- условная подгруппа: острый миелоидный лейкоз с мутантной СЕВРА,
• острый миелоидный лейкоз с изменениями, связанными с миелодисплазией,
• миелоидные неоплазии, ассоциированные с предшествующей терапией,
• другие варианты острого миелоидного лейкоза, неспецифицированные:
- острый миелоидный лейкоз с минимальной дифференцировкой,
- острый миелоидный лейкоз без созревания,
- острый миелоидный лейкоз с созреванием,
- острый миеломоноцитарный лейкоз,
- острый монобластный/острый моноцитарный лейкоз,
- острый эритролейкоз:
♦ чистый эритролейкоз,
♦ эритролейкоз, эритроидно/миелоидный,
- острый мегакариобластный лейкоз,
- острый базофильный лейкоз,
- острый панмиелоз с миелофиброзом,
Миелоидная пролиферация, относящаяся к синдрому Дауна:
- транзиторный аномальный миелопоэз,
- миелоидный лейкоз, ассоциированный с синдромом Дауна,
- властное новообразование из плазмоцитоидных дендритических клеток.
Острый лейкоз неопределенных линий:
- острый недифференцируемый лейкоз,
- острый лейкоз смешанного фенотипа с t (9; 22) (q34; q11,2); BCR-ABL1,
- острый лейкоз смешанного фенотипа с t (v; 11q23); и peaранжировкой MLL,
- острый лейкоз смешанного типа, В/миелоидный неспецифицированный,
- острый лейкоз смешанного фенотипа, Т/миелоидный неспецифицированный,
- условная подгруппа: лимфобластный лейкоз/лимфома из NK-клеток.
В-лимфобластный лейкоз/лимфома
В-лимфобластный лейкоз/лимфома, неспецифицированный
В-лимфобластный лейкоз/лимфома с повторяющимися генетическими аномалиями
В-лимфобластный лейкоз/лимфома с t (9:22) (q34; q11,2); BCR-ABL1
В-лимфобластный лейкоз/лимфома с t (v; 11q23); реаранжировка MLL
В-лимфобластный лейкоз/лимфома с t (12; 21) (p13; q22); TEL-AML1 (ETV6-RUNX1)
В-лимфобластный лейкоз/лимфома с гипердиплоидией
В-лимфобластный лейкоз/лимфома с гиподиплоидией
В-лимфобластный лейкоз/лимфома с t (5; 14) (q31; q32); IL3-IGH
В-лимфобластный лейкоз/лимфома с t (1; 19) (q23; p13,); E2A-PBX1 (TCF3-PBX1)
Читайте также: