
Хронический т-клеточный лейкоз из больших гранулярных лимфоцитов
Синдром, характеризующийся пролиферацией больших гранулярных лимфоцитов (БГЛ), в качестве самостоятельной нозологической формы был впервые описан в 1977 г. На тот момент точного терминологического определения заболеванию дано не было. R. McKenna и соавт. описали хроническое лимфопролиферативное заболевание с необычными клиническими, морфологическими, ультраструктурными и иммунологическими характеристиками.
За последние три десятилетия в литературе это заболевание называли по-разному: Ту-лимфопролиферативное заболевание, хронический Т-клеточный лимфоцитоз с нейтропенией, Т-клеточный хронический лимфолейкоз (Т-ХЛЛ), Т8+ Т-ХЛЛ. Все эти термины в той или иной степени отражали морфологические, иммунологические и функциональные характеристики опухолевых клеток. Так, в работе J. Brouet с соавт. лейкоз из больших гранулярных лимфоцитов охарактеризован как Т-клеточный хронический лимфолейкоз.
Одиннадцать случаев, описанных авторами, как показал последующий анализ, включали двух пациентов с Т-клеточным пролимфоцитарным лейкозом и девять с лейкозом из больших гранулярных лимфоцитов. По современным представлениям, существование Т-клеточного хронического лимфолейкоза оспаривается. Лишь в немногих исследованиях он охарактеризован как отдельное заболевание. J. Hoyer и соавт., опираясь в основном на морфологические критерии, описали 25 случаев Т-ХЛЛ. Следует ли эти случаи относить к Т-ХЛЛ или мелкоклеточному варианту Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ), в большей степени является предметом семантических, а не терминологических дискуссий.
Е. Matutes и соавт., изучив электронно-микроскопические, цитогенетические, иммунологические и клинические особенности Т-ХЛЛ у этой группы больных, небезосновательно относят его к мелкоклеточному варианту Т-ПЛЛ. Таким образом, данные литературы, посвященной Т-клеточным лимфопролиферативным заболеваниям, следует интерпретировать с большой осторожностью, учитывая весь спектр новых методов обследования и современные классификационные подходы.
Выделению лейкоза из больших гранулярных лимфоцитов в отдельную нозологическую форму немало способствовало изучение функции и морфологии различных популяций Т-лимфоцитов. Большие гранулярные лимфоциты составляют 5—20 % от общего количества лимфоцитов крови (в абсолютных значениях от 250 до 450 в 1 мкл крови). Это клетки диаметром 12—15 мкм, с умеренным или широким ободком цитоплазмы слабобазофильного цвета, содержащей нежные или более грубые (плотные) азурофильные гранулы, размер и количество которых значительно варьируют. Ядра клеток округлые или овальные, располагаются в центре или несколько эксцентрично. Хроматин ядер конденсированный, ядрышки не просматриваются.
Соответственно двум типам больших гранулярных лимфоцитов выделяют 2 варианта лейкоза из БЛГ (ЛБГЛ): из NK-клеток (CD3-) и цитотоксических Т-клеток (CD3+). NK-ЛБГЛ встречается значительно реже, чем Т-ЛБГЛ, в основном в азиатском регионе, характеризуется агрессивным течением с поражением лимфатических узлов, печени, селезенки и наличием В-симптомов.
Подавляющее большинство случаев лейкоза из больших гранулярных лимфоцитов представлено его Т-клеточным вариантом. Болеют в основном люди пожилого возраста, несколько чаще женщины. Т-клеточный лейкоза из больших гранулярных лимфоцитов (Т-ЛБГЛ) характеризуется доброкачественным клиническим течением и нередко протекает бессимптомно. Диагноз может быть поставлен при рутинном клинико-лабораторном обследовании. К диагностическим критериям Т-клеточного лейкоза из больших гранулярных лимфоцитов относятся увеличение абсолютного количества больших гранулярных лимфоцитов более 2 • 10 9 /л с CD3+CD8+-иммунофенотипом в течение 6 мес и более, нейтропения и/или анемия.
Известны случаи Т-клеточного лейкоза из больших гранулярных лимфоцитов с меньшим содержанием больших гранулярных лимфоцитов. У большинства пациентов симптомы заболевания связаны с рецидивирующими инфекциями на фоне нейтропении. Приблизительно у 20 % больных развитию Т-клеточного лейкоза из больших гранулярных лимфоцитов (Т-ЛБГЛ) предшествует длительный анамнез ревматоидного артрита. В этих случаях при физикальном обследовании определяется увеличение селезенки, обычно выступающей менее чем на 10 см из-под края реберной дуги. У половины больных обнаруживают спленомегалию, менее чем у 20 % пациентов манифестация заболевания проявляется кожными поражениями в виде папулезной сыпи. В противоположность В-ХЛЛ лимфаденопатия является достаточно редким симптомом (менее 5 % от всех случаев).
В клиническом анализе крови определяется нормальное или незначительно повышенное количество лейкоцитов (5—30•10 9 /л) с абсолютным лимфоцитозом (>5•109/л), причем клетки с характерными морфологическими особенностями больших гранулярных лимфоцитов. В подавляющем большинстве случаев количество нейтрофилов составляет менее 1,5•109/л. Анемия и тромбоцитопения выявляются приблизительно у 20 % больных.
В таблице представлены клинические данные 10 больных с Т-клеточным лейкозом из больших гранулярных лимфоцитов, которые находятся под нашим наблюдением.
Опухолевые клетки при Т-клеточном лейкозе из больших гранулярных лимфоцитов имеют характеристики антигенактивированных цитотоксических Т-лимфоцитов, однако их антигенная специфичность точно не установлена. По данным некоторых исследователей, в сыворотке больных Т-клеточным лейкозом из больших гранулярных лимфоцитов обнаружены антитела к HTLV-I/II, но убедительных свидетельств клональной интеграции ретровируса в геном лейкозных клеток не получено. Одним из основных механизмов персистенции лимфоцитов при Т-клеточном лейкозе из больших гранулярных лимфоцитов является нарушение регуляции апоптоза.
Нормальные цитотоксические Т-лимфоциты, распознавая вирусные пептиды при участии главного комплекса гистосовместимости, имеют высокий уровень экспресии Fas/CD95 и Fas/CD95-лиганда. Инфицированные вирусом клетки-мишени затем элиминируются путем Fas-зависимого апоптоза. Fas-лиганд относится к семейству белков фактора некроза опухоли и индуцирует апоптоз путем связывания со своим рецептором, который также известен как APO-I или CD95. В уничтожении антигенактивированных цитотоксических Т-лимфоцитов задействован тот же самый механизм. Показано, что лимфоциты при Т-ЛБГЛ также имеют высокий уровень экспрессии Fas и Fas-лиганда и их накопление обусловлено нарушением Fas-зависимого апоптоза. Устойчивая экспрессия и экскреция Fas-лиганда Т-БГЛ может являться одним из возможных механизмов развития нейтропении, особенно при отсутствии четкой корреляции между степенью нейтропении и объемом поражения костного мозга.
В норме нейтрофилы экспрессируют CD95 и подвергаются Fas-зависимому апоптозу, поэтому можно предположить, что избыточная секреция Fas-лиганда опухолевыми БГЛ может быть одной из причин нейтропении при Т-клеточном лейкозе из больших гранулярных лимфоцитов (Т-ЛБГЛ).
Важной отличительной чертой Т-клеточного лейкоза из больших гранулярных лимфоцитов (Т-ЛБГЛ) является ассоциация с другими заболеваниями. По данным ряда авторов, около 20—30 % больных Т-ЛБГЛ имеют серологические и клинические признаки ревматоидного артрита.
Аннотация научной статьи по фундаментальной медицине, автор научной работы — Доронин В. А., Никитин Е. А., Сидорова Ю. В., Пивнчк А. В.
Клинико-лабораторная картина лейкоза из больших гранулярных лимфоцитов (ЛБГЛ) нейт ропенин, спленомегалия, ассоциации с ревматоидным артиртом (РА) часто сходна с таковой при синдроме Фелти. Цель. Сравнение клинико-гематолоптческих, иммунофенотипических и иммуногенетически.', особенностей больных с синдромом Фелти и ЛБГЛ с и без РА. Материалы и методы. Представлена клинико-лабораторная характеристика 10 больных с ЛБГЛ. Иммунофеиотипическое исследование лимфоцитов проводилось с помошью моноклональных антител CD8-PE и CD3-FITC/CDI6-PE (двойная метка) (Caliag. USA) методом проточной цитометрии (Partec, Daco). При исследовании клональной перестройки генов гамма-цепи Т-клеточного рецептора использовался ПЦР анализ конформаиионного полиморфизма одноцепочечных фрагментов ДНК в растворе с низкой ионной силой. Результаты. ЛБГА является отдельной нозологической формой. Заболевание часто ассоциирует с РА (около 20%). Пациенты с ЛБГЛ + РА и пациенты с синдромом Фелти имеют схожие иммуногенетические характеристики, которые не выяатяются у больных с ЛБГЛ без РА. Заключение. Морфологическое, иммунофеиотипическое и молекулярногенетическое исследования являются крайне важными и высокорепрезентативными методами в дифференциальной диагностике нейтропении при РА. Синдром Фелти и ЛБГЛ являются вариантами синдрома, характеризующегося нейтропенией, спленомегалией, наличием РА и пролиферацией больших гранулярных лимфоцитов. Т-БГЛ распознают чужеродный антиген в комплексе с молекулой главного комплекса гистосовместимости (МНС) 1 класса через Т-клеточный рецептор. Гены Т-клеточного рецептора в этих клетках перестроены. В западных странах ЛБГД в подавляющем большинстве случаев возникает именно из Т-БГЛ. Около 80-90% БГЛ являются естественными киллерами (НК-клетки). НК-БГЛ принципиально отличаются от Т-БГЛ (таблица 1). Опухоли из НК-клеток в западных странах встречаются крайне редко, имеют совершенно иную клинику и не сочетаются с РА. В таблице 1 приведена сравнительная характеристика двух популяций БГЛ. Ключевые слова: лейкоз из больших гранулярных лимфоцитов , ревматоидный
Похожие темы научных работ по фундаментальной медицине , автор научной работы — Доронин В. А., Никитин Е. А., Сидорова Ю. В., Пивнчк А. В.
Neutropenia in rheumatoid arthritis and large granular lymphocyte leucosis
Objective. Pts with chronic clonal proliferation of large granular lymphocytes (LGL leukemia) often have neutropenia, splenomegaly, and rheumatoid arthritis (RA), thereby resembling the manifestations observed in pts with Felty’s syndrome. The present study sought to indicate that pts with these disorders represent two distinct subsets. We compare clinical, hematological, immunophenotiping and immunogenetic features in Felty’s syndrome pts with and without the LGL leukemia. Material and methods 10 pts with T-LGL leukemia were studied. Surface phenotype was estimated using monoclonal antibodies CD8-PE and CD3-FITC/CD16-PE (two-color) (Caltag, USA) by the flow cytometric analysis (Partec, Daco). Analysis of TCR gene rearrangement was performed by using PCR-LIS SSCP (low ionic strength single strand conformational polymorphism). Comparison with Felty s syndrome and RA pts based on the review of literature. Results. LGL leukemia is a distinct clinicopathologic entity often associated with RA. LGL leukemia pts with RA showed the same immunogenetis associations seen in RA/Felty’s syndrome, while LGL leukemia pts without arthritis did not. Conclusion. Hematologic, immunophenotyping and molecular genetic analysis are very important and highly representative tools in differential diagnosis of neutropenia in RA, and propose that Felty’s syndrome and LGL leukemia represent different variants of broader syndrome comprising RA, neutropenia, LGL expansions, and splenomegaly.
УДК: 616.155.3-02: 616.72-002.77
НЕЙТРОПЕНИЯ ПРИ РЕВМАТОИДНОМ АРТРИТЕ И ЛЕЙКОЗ ИЗ БОЛЬШИХ ГРАНУЛЯРНЫХ ЛИМФОЦИТОВ
В.А. Доронин1, Е.А. Никитин2, Ю.В. Сидорова2, A.B. Пивник1
Клинико-лабораторная картина лейкоза из больших гранулярных лимфоцитов (ЛБГЛ) - нейт ропенин, спленомегалия, ассоциация с ревматоидным артиртом (РА) - часто сходна с таковой при синдроме Фелти.
Цель. Сравнение клинико-гематолоптческих, иммунофенотипических и иммуногенетически.', особенностей больных с синдромом Фелти и ЛБГЛ с и без РА.
Материалы и методы. Представлена клинико-лабораторная характеристика 10 больных с ЛБГЛ. Иммунофеиотипическое исследование лимфоцитов проводилось с помошью моноклональных антител CD8-PE и CD3-FITC/CDI6-PE (двойная метка) (Caliag. USA) методом проточной цитометрии (Partee, Daco). При исследовании клональной перестройки генов гамма-цепи Т-клеточного рецептора использовался ПЦР анализ конформаиионного полиморфизма одноцепочечных фрагментов ДНК в растворе с низкой ионной силой.
Результаты. ЛБГА является отдельной нозологической формой. Заболевание часто ассоциирует с РА (около 20%). Пациенты с ЛБГЛ + РА и пациенты с синдромом Фелти имеют схожие иммуногенетические характеристики , которые не выяатяются у больных с ЛБГЛ без РА. Заключение. Морфологическое, иммунофенотипическое и молекулярногенетическое исследования я ал я юте я крайне важными и высокорепрезентативными методами в дифференциальной диагностике нейтропении при РА. Синдром Фелти и ЛБГЛ являются вариантами синдрома, характеризующегося нейтропенией, спленомегалией, наличием РА и пролиферацией больших гранулярных лимфоцитов.
Ключевые слова: лейкоз из больших гранулярных лимфоцитов, ревматоидный артрит, синдром Фелти.
Нейтропения при ревматоидном артрите (РА) является актуальной проблемой. Она осложняет течение РА, представляет дифференциально-диагностическую проблему и требует особой терапевтической тактики. Механизмы нейтропении при РА до сих пор изучены слабо. Они носят комплексный характер. У большей части больных с нейтропенией и РА обнаруживаются антитела против гранулоци-тарно-макрофагального колоние-стимулируюшего фактора (Г-КСФ) или антигенов на поверхности нейтрофилов [10,
16]. Возможно подавление продукции нейтрофилов в костном мозге цитотоксическими клетками [7]. Повреждение нейтрофилов может осуществляться иммунными комплексами [3, 2, 19]. Обсуждаются и не иммунные механизмы развития нейтропении: индукция апоптоза растворимым FAS-лигандом, индукция нечувствительности миелоидных предшественников к Г-КСФ [7]. Описаны случаи фагоцитоза нейтрофилов макрофагами костного мозга при синдроме Фелти [11]. Ранее к причинам нейтропении при данном синдроме относили секвестрацию нейтрофилов в селезенке.
В последние годы стало известно, что одной из причин нейтропении при РА может быть Т-клеточный лейкоз из больших гранулярных лимфоцитов (Т-ЛБГЛ). В настоящее время морфология, функция и иммунологические характеристики больших гранулярных лимфоцитов (БГЛ) изучены подробно [21]. Они составляют 5-20% от общего количества лимфоцитов крови (в абсолютных значениях -от 250 до 450 в 1 мкл крови). Популяция БГЛ гетерогенна по функциональным и иммунофенотипическим характеристикам. Около 10-20% БГЛ относятся к цитотоксическим Т-лимфоцитам (Т-БГЛ) с иммунофенотипом CD3+.CD8+.
* - Городской гематологический центр ГКБ’им. С.П.Боткина, Москва
** - Гематологический Научный Центр РАМН, Москва.
Т-БГЛ распознают чужеродный антиген в комплексе с молекулой главного комплекса гистосовместимости (МНС) 1 класса через Т-клеточный рецептор. Гены Т-клеточного рецептора в этих клетках перестроены. В западных странах ЛБГД в подавляющем большинстве случаев возникает именно из Т-БГЛ.
Около 80-90% БГЛ являются естественными киллерами (НК-клетки). НК-БГЛ принципиально отличаются от Т-БГЛ (таблица 1). Опухоли из НК-клеток в западных странах встречаются крайне редко, имеют совершенно иную клинику и не сочетаются с РА. В таблице 1 приведена сравнительная характеристика двух популяций БГЛ.
СРАВНИТЕЛЬНАЯ ХАРАКТЕРИСТИКА НК- И Т-БГЛ
Морфология Большие гранулярные лимфоциты
Антигенная специфичность распознавания Есть Нет
Гены Т-клеточного рецептора Перестроены Не перестроены
Распознавание мишени Через Т- клеточный рецептор Через рецепторы НК-клеток
Уничтожение мишени Перфорины, гранзимы, индукция апоптоза в клетке мишени
Иммунофенотип CD3+, CD8+ г ■ CD3-, CD8-, CD16+, CD56+
С момента первого описания в 1977г Т-ЛБГЛ имел разные названия: Т-хронический лимфолейкоз, Т8-гипер-лейкоцитоз, Т -лимфопролиферативное заболевание, Т-клеточный лимфоцитоз с нейтропенией [18]. В начале 90х годов, после доказательства клональной природы этой болезни, Т-ЛБГЛ был выделен в отдельную нозологическую форму [14]. Диагностические критерии Т-ЛБГЛ таковы:
1) наличие лимфоцитоза за счет больших гранулярных лимфоцитов > 2000/мкл, сохраняющегося >6 мес:
2) иммунофенотип БГЛ (СЭЗ+, С08+);
3) нейтропения и/или другая цитопения, сохраняющаяся > 6 мес;
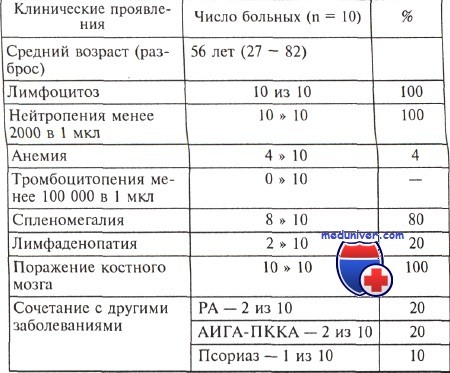
Наиболее характерными клиническими проявлениями Т-ЛБГЛ являются лимфоцитоз, нейтропения и сплено-мегалия. В таблице 2 представлены клинические данные по 10 больным с Т-ЛБГЛ, которые находятся под нашим наблюдением.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ЛЕЙКОЗА ИЗ БОЛЬШИХ ГРАНУЛИРОВАННЫХ ЛИМФОЦИТОВ
Клинические проявления Число больных (п=10) %
Средний возраст ,годы 56 (27 - 82)
Лимфоцитоз 10 100
Нейтропения Не можете найти то, что вам нужно? Попробуйте сервис подбора литературы.
drome: autoimmune neutropenia or immune-complex-mediated disease? Rheumatol. Int., 1985, 5 (6), 253-8.
3. Breedveld F.C., Lafeber G.J., de Vries E. et al. Immune com-
plexes and the pathogenesis of neutropenia in Felty’s syndrome. Ann.Rheum.Dis., 1986, 45(8), 696-702.
4. Bowman S.J., Corrigall V., Panayi G.S., Lanchbury J.S.
Hematologic and cytofluorographic analysis of patients with Felty's syndrome. A hypothesis that a discrete event leads to large granular lymphocyte expansions in this condition.. Arthr. Rheum., 1995,38(9), 1252-9.
5. Bowman S.J., Sivakumaran М., Snowden N. et al. The large
granular lymphocyte syndrome with rheumatoid arthritis. Immunogenetic evidence for a broader definition of Felty's syndrome. Arthr. Rheum., 1994,37(9), 1326-30.
6. Coakley G., Brooks D., Iqbal M. et al. Major histocom-
patility complex haplotypic associations in Felty's syndrome and large granular lymphocyte syndrome are secondary to allelic association with HLA-DRB1 *0401. Rheumatol., 2000, 39, 393-398
7. Coakley G., Iqbal М., Brooks D. et al. CD8+, CD57+ T
cells from healthy elderly subjects suppress neutrophil development in vitro: implications for the neutropenia of Felty's and large granular lymphocyte syndromes. Arthr.Rheum.,2000,43(4),834-43.
8. Dhodapkar M.V., Li C.Y., Lust J.A. et al. Clinical spectrum
of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood. 1994, 84(5), 1620-7.
9. Gabor E.P., Mishalani S., Lee S. Rapid response to
cyclosporine therapy and sustained, remission in large granular lymphocyte leukemia. Blood.,1996,87(3),1199-200.
10. Hellmich B., Csemok E., Schatz H. et al. Autoantibodies against granulocyte colony-stimulating factor in Felty's syndrome and neutropenic systemic lupus ervthematosus. Arthr. Rheum., 2002,46(9),2384-91.
11. Kumakura S., Kobayashi S., Ishikura H. Neutrophil phagocytosis in Feltv's syndrome. Am .J. Med., 2001,111(7),579-80.
12. Lamy T., LePrise P.Y., Amiot L. et al. Response to granulocyte-macrophage colony-stimulating factor (GM-CSF) but not to G-CSF in a case of agranulocytosis associated with large granular lymphocyte (LGL) leukemia. Blood,1995,85(11),3352-3.
13. Lamy T.,Loughran TP. Large Granular lymphocyte leukemia. Cancer Control., 1998,5(1),25-33.
14. Loughran T.P. Jr.Clonal diseases of large granular lymphocytes. Review. Blood, 1993,82(1), 1-14.
15. Loughran T.P.Jr.,Kidd P.G., Starkebaum G. Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood, 1994,84(7),2164-70.
16. Maher G.M., Hartman K.R. Detection of antineutrophil autoantibodies by flow cytometry: use of unfixed neutrophils as antigenic targets. J.Clin.Lab.Anal., 1993,7(6),334-40.
17. Matutes E. T-cell lymphoproliferative disorders. Classification, clinical and laboratory aspects. Ed: A. Polliack. Harwood Academic Publishers., 1999, 23.
18. McKenna R.W., Parkin J., Kersey J.H. et al. Chronic lymphoproliferative disorder with unusual clinical, morphological, ultrastructural and membrane surface marker characteristics. Am. J. Med.. 1977,62(4),588-96.
19. Petersen J., Wiik A. Lack of evidence for granulocyte specific membrane-directed autoantibodies in neutropenic cases of rheumatoid arthritis and in autoimmune neutropenia. Acta Pathol. Microbiol. Immunol. Scand. [C]., 1983,91(1),15-22.
20. Starkebaum G., Loughran T.P. Jr, Gaur L.K. et al. Immunogenetic similarities between patiente with Felty's syndrome and those with clonal expansions of laige granular lymphocytes in rheumatoid arthritis. Arthr.Rheum.,1997,40(4),624-6.
21. Timonen T., Saksela E., Ranki A., Hayry P. Fractionation,
morphological and functional characterization of effector cells responsible for human natural killer activity against cell-line targets. Cell Immunol.,1979,48(1),133-48.
22. Yoe J.,Gause B.L.,Curti B.D. et al. Development of rheumatoid arthritis after treatment of large granular lymphocyte leukemia with deoxycoformycin. Am.J. Hematol.,1998,57(3) ,253-257.
Neutropenia in rheumatoid arthritis and large granular lymphocyte leucosis
V.A. Domnin, E.A. Nikitin, J.V. Sidorova, A. V Pivnik
Objective. Pts with chronic clonal proliferation of large granular lymphocytes (LGL leukemia) often have neutropenia, splenomegaly, and rheumatoid arthritis (RA), thereby resembling the manifestations observed in pts with Felty’s syndrome. The present study sought to indicate that pts with these disorders represent two distinct subsets. We compare clinical, hematological, immunophenotiping and immunogenetic features in Felty’s syndrome pts with and without the LGL leukemia.
Material and methods 10 pts with T-LGL leukemia were studied. Surface phenotype was estimated using monoclonal antibodies CD8-PE and CD3-FITC/CD16-PE (two-color) (Caltag, USA) by the flow cytometric analysis (Partec, Daco). Analysis of TCR gene rearrangenient was performed by using PCR-LIS SSCP (low ionic strength single strand conformational polymorphism). Comparison with Felty s syndrome and RA pts based on the review of literature.
Results. LGL leukemia is a distinct clinicopathologic entity often associated with RA. LGL leukemia pts with RA showed the same immunogenetis associations seen in RA/Felty’s syndrome, while LGL leukemia pts without arthritis did not.
Conclusion. Hematologic, immunophenotyping and molecular genetic analysis are very important and highly representative tools in differential diagnosis of neutropenia in RA, and propose that Felty’s syndrome and LGL leukemia represent different variants of broader syndrome comprising RA, neutropenia, LGL expansions, and splenomegaly.
Key words: Large granular lymphocytes leukemia, rheumatoid arthritis, Felty's syndrome.

Представлена клинико-гематологическая и иммунофенотипическая характеристика Т-клеточного варианта лейкоза из больших гранулосодержащих лимфоцитов. Выделены дифференциально-диагностические признаки и приведены протоколы лечения.
Синдром, характеризующийся пролиферацией больших гранулосодержащих лимфоцитов (БГЛ), был впервые описан в 1977 г. Опухолевая природа этого синдрома долгое время вызывала сомнения в связи с отсутствием надежных способов доказательства клональности. Проведение иммунофенотипических и молекулярно-генетических методов исследования для определения Т-клеточной клональности позволило охарактеризовать своеобразную лимфатическую опухоль и выделить ее среди многочисленных случаев Т-клеточного лимфоцитоза [1,2].
На протяжении последних лет в Украине наблюдается рост числа больных с хроническими лимфопролиферативными заболеваниями. Клинические проявления хронического лейкоза из больших гранулосодержащих лимфоцитов чаще всего сопровождаются гранулоцитопенией. Опухолевые клетки демонстрируют своеобразную морфологию, давшую название заболеванию. Характерен умеренный лимфатический лейкоцитоз, часто с абсолютной нейтропенией. Не существует единства мнений в вопросе о том, при каком количестве лимфоцитов может быть установлен диагноз лейкоза из больших гранулосодержащих лимфоцитов. Полагают, что уровень лимфоцитов менее 5х109/л свидетельствует о реактивном лимфоцитозе, а более 5х109/л указывает на наличие Т-клеточного лейкоза из БГЛ. В большинстве случаев при лейкозе из БГЛ при умеренном лимфоцитозе (
8х109/л) определяется более 2х109/л БГЛ [3].
Не выявлено также связи между выраженностью нейтропении и степенью поражения костного мозга. В связи с частым обнаружением в крови (по данным различных авторов) антинейтрофильных антител не исключена аутоиммунная природа нейтропении [4].
Выделение лейкоза из больших гранулосодержащих лимфоцитов в отдельную нозологическую форму немало способствовало изучению функции и морфологии Т-лимфоцитов. Иммунофенотипически БГЛ гетерогенны. 80-90% всех больших гранулосодержащих лимфоцитов являются естественными киллерами (NK-клетки). Гены Т-клеточного рецептора в этих клетках не перестроены. Они не экспрессируют на своей поверхности молекулы, ассоциированные с Т-клеточным рецептором (CD3, CD8, CD4), и имеют иммунофенотип CD56 + , CD3 - , CD8 - , CD16 + .
10-20% БГЛ – это цитотоксические Т-лимфоциты, имеющие иммунофенотип CD57 + , CD3 + , CD8 + и CD16 + [1,3].
Многие аутоиммунные заболевания сопровождаются пролиферацией NK и Т-клеток (CD8 + ), особенно ревматоидный полиартрит, при этом зачастую развивается нейтропения различной степени выраженности и спленомегалия (синдром Фелти). Относительные и абсолютные лимфоцитозы могут быть следствием многих перенесенных заболеваний, в том числе инфекционных, как вирусной (аденовирусная инфекция, грипп, цитомегаловирусная инфекция, вирусные гепатиты), так и невирусной природы (токсоплазмоз, туберкулез, сифилис, малярия и др.). Как правило, эти лимфоцитозы поликлональны, содержание циркулирующих в крови лимфоцитов и их субпопуляционный состав нормализуется через 1-5 месяцев после болезни.
Иногда наблюдается моноклональный лимфоцитоз, дифференциальная диагностика которого может вызывать некоторые затруднения. Яркий пример – инфекционный мононуклеоз, при котором в периферической крови наблюдается абсолютный лимфоцитоз с преобладанием активированных Т-лимфоцитов с фенотипом Т-клеток – хелперов (CD3 + CD4 + ) [4, 5].
Таким образом, дифференциальная диагностика хронических лейкозов из больших гранулосодержащих лимфоцитов и реактивных лимфоцитозов вызывает большие затруднения и невозможна без проведения иммунофенотипических исследований.
Результаты и обсуждение
В данной статье мы хотели бы рассмотреть вопросы диагностики и лечения хронического лимфолейкоза из больших гранулосодержащих лимфоцитов на примере клинического случая.
Больная П., 1947 года рождения, была направлена в гематологическое отделение в связи с анемией, лейкопенией и тромбоцитопенией на обследование. При осмотре не было выявлено увеличения лимфатических узлов, селезенки и печени. После проведения иммуноцитохимических исследований у больной не был установлен диагноз хронического лимфопролиферативного заболевания (не установлена моноклоновость процесса).
Больная находилась (на протяжении 4 лет) под наблюдением гематолога в поликлинике. В связи с низкими показателями количества лейкоцитов и эритроцитов, больная периодически принимала преднизолон. Частые воспалительные заболевания, что сопровождались повышением температуры тела до 38-39ºС, появление органомегалии (сплено- и гепатомегалии), а также изменения в анализе крови (лейкопения с лимфоцитозом) привели к вторичному обследованию больной в гематологическом отделении. На этот раз отмечалась бледность кожных покровов без геморрагических проявлений и увеличения лимфатических узлов. Обращала на себя внимание спленомегалия (на 5-6 см выступала из-под нижнего края реберной дуги) и гепатомегалия (на 2-3 см пальпировалась ниже реберной дуги).
В анализе крови: Hb 100г/л, эр. 2,95х1012/л, тр. 31,9х109/л, л. 2,5х109/л, с. 9%, лимф. 75%, мон. 11%, э. 4%, б. 1%, СОЭ 53мм/час. В миелограмме количество лимфоцитов составило 60%. Морфологически лимфоциты выглядели крупнее типичных малых лимфоцитов. Ядро имело круглую или овальную форму. По сравнению с сегментоядерным нейтрофилом хроматин менее конденсирован. Широкая светло-голубая цитоплазма, содержащая азурофильные гранулы. При проведении цитохимических исследований определялась яркая реакция на кислую фосфатазу и мелкогранулярная PAS-реакция в субстратных клетках. Иммунофенотипически мононуклеары периферической крови экспрессировали: CD3 80%, CD2 55%, CD8 65%, CD7 50%, CD4 22%, NK 59%, CD20 7%, CD19 5%.
На основании проведенных исследований, больной установлен диагноз хронического лимфолейкоза из больших гранулосодержащих лимфоцитов. После проведения 2-х курсов полихимиотерапии по схеме CHOP (циклофосфан, доксорубицин, винкристин, преднизолон) исчезла органомегалия и значительно улучшились показатели крови. Так, в анализе крови: эр. 4,0х1012/л, Hb 115 г/л, тр. 182,0х109/л, л. 4,2х109/л, п. 2%, с. 65%, лимф. 27%, мон. 4%, э. 2%, СОЭ 15 мм/час. В настоящее время больная наблюдается гематологом в поликлинике без лечения.
Данный случай полностью соответствует диагностическим критериям Т-клеточного лейкоза из больших гранулосодержащих лимфоцитов, к которому относится повышение абсолютного количества БГЛ, цитопения и иммунофенотипически преобладание в крови клеток, экспрессирующих CD3 и CD8.
В заключении отметим, что в настоящее время стандарты терапии больных с этой редкой патологией не разработаны. Показаниями для начала терапии являются нейтропения (менее 500 клеток/мкл), осложненная частыми инфекциями, глубокая анемия, выраженная спленомегалия. Медиана выживаемости при Т-клеточном варианте БГЛ превышает 10 лет. В связи с длительным хроническим течением заболевания оправдана выжидательная тактика, стремление, по возможности, избегать миелодепрессивной химиотерапии. В случае глубокой нейтропении применяют метотрексат в виде монотерапии или в сочетании с преднизолоном, циклоспорин, ростовые факторы [6].
По данным T.P. Loughran и соавт., монотерапия преднизолоном дает эффект только во время лечения: с уменьшением дозы уровень гранулоцитов снижается, а лимфоцитоз возрастает. Наконец, при выраженном прогрессировании заболевания применяют полихимиотерапию (СНОР), флюдарабин, метотрексат в сочетании с преднизолоном. Что касается диагностики Т-клеточного лейкоза из БГЛ требуется морфологическое изучение мазка периферической крови, а в трудных для диагностики случаях рекомендуется определение Т-клеточной клональности [7].
Выводы
- Диагностика Т-клеточного лейкоза из больших гранулосодержащих лимфоцитов должна включать иммуноцитохимические методы с использованием широкой панели моноклональных антител.
- Показаниями для назначения специфической терапии является нейтропения, которая осложняется частыми инфекциями, глубокой анемией и спленомегалией.
1 Родионова И.А., 1 Скрипниченко С.В., 2 Булавина В.П.
1 Национальный медицинский университет им. А.А. Богомольца,
2 Киевская городская клиническая больница № 9
Читайте также: