
Генетические маркеры лимфопролиферативного заболевания
Аутоиммунный лимфопролиферативный синдром – передающееся по наследству патологическое состояние. Принадлежит к категории гетерогенных. Есть два механизма наследования: аутосомный доминантный и рецессивный. В редких случаях причина – соматические мутации. Лимфопролиферативный синдром может быть приобретенным.
История и факты
Впервые первоначальный х-сцепленный лимфопролиферативный синдром мальчиков был официально признан и оформлен в науке в 1967 году. С 1976 его причисляют к первичному иммунодефициту. Внимание ученых к патологическому состоянию приковано с последних десятилетий прошлого столетия. Уже тогда было выявлено, что базой развития заболевания становится неправильный лимфоцитный апоптоз.

Выявляя особенности аутоиммунного лимфопролиферативного синдрома, ученые установили, что всем больным свойственна неправильная экспрессия рецепторов мембран fasl, CD95. Именно этот нюанс определяет генетически объясняющуюся способность клеток умирать. Патологическое состояние развивается в случае генной мутации, влияющей на апоптоз.
Биология и анатомия: как все происходит?
В медицинской пропедевтике лимфопролиферативный синдром принято делить на несколько разновидностей. Для классификации учитывают особенности генетических отличий конкретного случая. Генные мутации могут затрагивать восьмую и десятую каспазы, CD95, CD178. В то же время стоит отметить, что не существует общепризнанной официальной классификации случаев на группы.
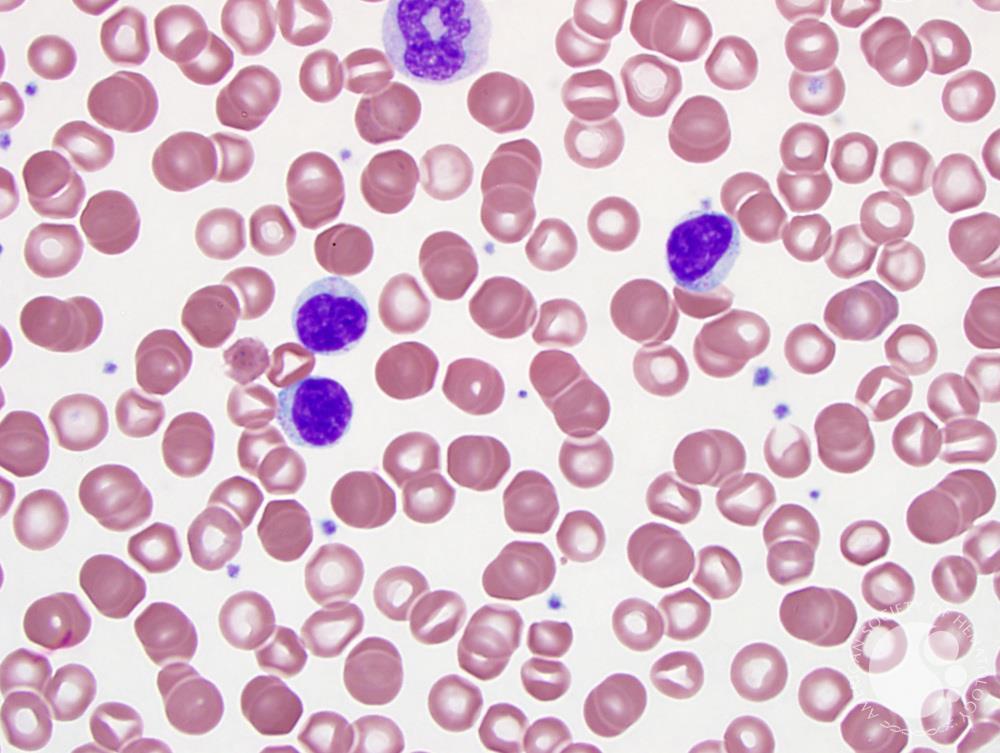
Особенности проявления
Симптомы заболевания исключительно разнообразны. Обычно х-сцепленный лимфопролиферативный синдром выявляют на первых годах жизни, несколько реже – в более старшем возрасте (до пятнадцатилетнего). Ключевой симптом – пролиферация лимфоидной ткани, провоцирующая спленомегалию, лимфаденопатию. Явлениям присущ хронический характер течения. Одновременно больной страдает от проявлений аутоиммунного дисбаланса. Анализы помогают выявить аутоиммунную цитопению. Она возможна в форме нейтро-, тромбоцитопении, анемии. Несколько реже цитопения появляется ранее пролиферации лимфоидных тканей.
Х-сцепленный лимфопролиферативный синдром провоцирует нарушения в работе кроветворной, кровеносной систем. Как правило, фиксируется гепатит аутоиммунной природы. Многие страдают от экземы, гломерулонефрита. Пациентам свойственны увеит, тиреоидит. Приблизительно у каждого десятого со временем формируется лимфома из клеток типа В.
Клинические проявления
Лимфопролиферативный синдром у детей имеет ряд типовых признаков. Наиболее яркий – лимфопролиферация. Процессу присущ доброкачественный характер, патологическое состояние хроническое. Обычно формируется уже в раннем детстве, иногда устанавливается у годовалых малышей. Состояние сохраняется от полугода и более. Вместе с тем наблюдается персистирующее разрастание лимфоузлов периферической лимфосистемы. Для постановки диагноза необходимо выявить такие процессы в трех группах узлов или большем количестве. Узлы плотные, с расположенными поблизости тканями не спаяны. У многих анализы помогают выявить гепатоспленомегалию.
Х-сцепленный лимфопролиферативный синдром у мальчиков проявляет себя аутоиммунными признаками. Классический вариант – анемия, нейтро-, тромбоцитопения. Возможен васкулит. Нередки случаи артрита, гепатита. Больные склонны к увеиту, гломерулонефриту, тиреоидиту. Возможны некоторые другие болезни аутоиммунной природы.
Обратить внимание!
Лимфопролиферативный синдром сопряжен с высокой вероятностью развития злокачественного формирования. Область локализации процесса непредсказуема. Неправильно протекающий апоптоз, работа которого сопряжена с активностью рецепторов Fas, приводит к понижению контроля за процессами разрастания тканей. Растет способность выживать у клеток, переживших патологическую трансформацию. В норме указанный ген – это угнетающий развитие компонентов опухолей фактор.
Чаще заболевание сопровождается формированием лимфом типа В, Т. Кроме того, высока вероятность раковых процессов в молочной железе, кишечном тракте, органах дыхания. Миело-лимфопролиферативный синдром с высокой степенью вероятности может спровоцировать лимфогранулематоз.
При аутоиммунном заболевании пациент склонен к крапивнице, васкулиту. У некоторых отмечается замедленное развитие организма.
Уточнение диагноза
Лимфопролиферативный синдром диагностируют, если установлена не носящая злокачественный характер лимфаденопатия. Возможна спленомегалия. Диагноз ставят при комбинации этих двух явлений или присутствии любого из них, если длительность развития состояния – полгода и больше. При подозрении на диагноз необходимо направить пациента на анализы. В лабораторных условиях устанавливают сбой опосредованного лимфоцитного апоптоза, уточняют концентрацию клеточных структур CD4, CD8 Т: при содержании более 1% можно говорить о патологическом состоянии.
Особенности случая
Далее рассматривается форма заболевания, не связанная с нарушением генетики в период развития эмбриона. Нижеописанное касается приобретенной формы заболевания.
Лимфопролиферативный синдром – симптомокомплекс, который может сопровождать не только лимфолейкоз, протекающий по типовому сценарию, но и более редкие формы патологического состояния. Иногда его устанавливают при волосатоклеточным лейкозе, лимфатическом, в качестве осложнения которого – цитолиз. Известно, что комплекс симптомов может развиться на фоне медикаментозной терапии, облучения, влияния химических компонентов. Большое внимание в современной медицине привлекает посттрансплантационный лимфопролиферативный синдром, существенно ухудшающий прогнозы перенесшего операцию человека. В развитии синдрома, полученного не наследственным путем, наиболее сильно влияние ретровирусов.
Нюансы и распространенность
Медицинская статистика показывает, что преимущественный процент пациентов с лимфопролиферативным синдромом – люди старше пятидесятилетнего возраста. Изредка заболевание выявляется у тех, кто младше 25, но такие случаи единичны. Среди мужского пола частота встречаемости в среднем вдвое выше, нежели у женщин. Исходя из течения, говорят о доброкачественной форме, спленомегалической, опухолевой, склонной к быстрому прогрессу, затрагивающей костный мозг, брюшную полость. Также есть пролимфоцитарный тип.
Когда только начинает развиваться лимфопролиферативный синдром, внутренние болезни не беспокоят, человек чувствует себя удовлетворительно, отсутствуют активные жалобы. Некоторые отмечают слабость, склонность к простудам. Несколько активнее нормы функционируют потовые железы. Заболевание на этом этапе можно выявить в рамках профилактического обследования или на случайном осмотре. Основные признаки – ненормально крупные лимфоузлы, лимфоцитоз, повышение концентрации лейкоцитов в кровеносной системе.
Специфика симптоматики
При заболевании склонны к увеличению лимфоузлы на шее, в подмышечной ямке. Несколько позже, когда болезнь приобретает развернутую форму, отмечается увеличение прочих групп. Размеры сильно варьируются, как и консистенция: некоторых похожи на неплотное тесто, при исследовании болью не отзываются, между собой или с кожными покровами не сливаются. Для таких участков нехарактерно формирование язв или нагноений.
Когда болезнь приобретает развернутую форму, проявления становятся ярко выраженными, пациент ощущает себя слабым, резко понижается способность работать. У больного активно работают потовые железы, он теряет вес, страдает от жара. Лимфатические узлы существенно увеличены, что и привлекает внимание при первичном осмотре.
Осмотр больного: комплекс проявлений
При обследовании пациента заподозрить лимфопролиферативный синдром можно, если четко диагностируется лифмоаденопатия. У многих больных видна трансформация отдельных участков кожи: появляется инфильтрат, выявляются неспецифические пораженные участки. Если человек ранее страдал от кожных заболеваний, они обостряются в силу описываемого синдрома. Многих беспокоит эксфолиативная эритродермия. На фоне синдрома возможно развитие герпеса, крапивницы, дермита.
Для уточнения состояния необходимо направить больного на КТ, УЗИ. На лимфопролиферативный синдром указывает рост лимфоузлов в грудине, брюшной полости, при этом состояние не всегда сопровождается проявлениями компрессии. У пациента больше нормы селезенка, печень. Изучение слизистых пищеварительного тракта позволяет заметить лейкемическую инфильтрацию. Дополнительные проявления – язвы в желудке, кишечном тракте, кровотечения в этой области. Есть вероятность мальабсорбционного синдрома.
Прогресс состояния
При лимфопролиферативном синдроме возможно вовлечение дыхательной системы в патологические процессы. Лейкемическая инфильтрация может затронуть как верхние отделы, так и нижние пути прохождения воздуха. Больной кашляет, беспокоит одышка, возможно отхаркивание мокроты с кровянистыми включениями. Иногда устанавливают плеврит.
В ряде случаев описанный синдром провоцирует инфильтрацию почечной паренхимы. Такое состояние крайне редко проявляет себя типичной симптоматикой. Возможно распространение инфильтрата на ЦНС, что приводит к менингиту, некоторым формам энцефалита и параличу нервных структур, может стать причиной комы. При распространении инфильтрата на кавернозные тела больной страдает от продолжительной и провоцирующей боль эрекции, в медицине называемой приапизмом.
Лабораторные анализы
При подозрении на лимфопролиферативный синдром пациента направляют на исследование крови. Указанное состояние сопровождается ростом концентрации лимфоцитов, лейкоцитов. Возможна анемия.
Лабораторные анализы помогают диагностировать у пациента гемат-, протеинурию. Анализ на биохимию уточняет гипогаммаглобулинемию. В небольшом проценте случаев у пациентов устанавливают гипоальбуминемию. Гепатоцитный цитолиз указывает на себя гиперферментемией.
Иммунологическое исследование указывает на повышение концентрации в селезенке, кровеносной системе лимфоцитов, сбой баланса хелперов и супрессоров из числа лимфоцитов. Вместе с тем снижается концентрация IgG, IgA, IgM (для двух последних изменения особенно ярко выражены). Иммунофенотипирование – основание заключить, что лейкозные клеточные структуры – CD 5, 19, 20, 23 из класса В-лимфоцитов. Результаты цитогенетического анализа в 65 % случаев указывают на аномалии хромосом.

Что делать?
При лимфопролиферативном синдроме больному показано соблюдение разработанного врачом лечебного режима – программа выбирается индивидуально. Пациенту назначают цитостатические препараты. Особенно актуально это, если состояние здоровья быстро ухудшается, печень и селезенка, лимфоузлы стремительно увеличиваются. Цитостатики незаменимы при лейкемической инфильтрации волокон ЦНС, а также в случае, если процессы затрагивают органы вне кроветворной системы. Состояние указывает на себя сильной болью и сбоем функциональности систем и органов.
Если количество лейкоцитов в кровеносной системе неуклонно и быстро растет, показаны хлорбутин, спиробромин. Неплохую реакцию организма позволяют получить проспидин, циклофосфан. Иногда врачи рекомендуют остановиться на пафенциле. Если к этому есть специфические показания, могут назначить полихимиотерапию. В рамках такого курса цитостатические средства, влияющие на организм разными образами, комбинируют между собой.
Мероприятия и способы: как помочь пациенту?
При повышении содержания лейкоцитов до уровня 200*10 на 9/л рекомендован лимфоцитафарез. Если отдельные лимфоузлы резко и сильно увеличиваются, такие процессы выявлены в селезенке, если лимфаденопатия переходит в системную генерализованную форму, назначают лучевое лечение. При разрастании селезенки, не корректирующемся медикаментозными средствами и облучением, пациенту рекомендована спленэктомия. Ее необходимо пройти, если часты инфаркты этого органа, а также при заболевании, сопровождающемся выраженной спленомегалией, определенными формами лейкоцитозома, лейкоза. Спленэктомия незаменима при гранулоцито-, эритро-, тромбоцитопении, анемии аутоиммунного типа, тромбоцитопении, которую не удается регулировать глюкокортикоидами.
Если гормональные соединения показывают выраженный эффект при тромбоцитопении, если установлена гемолитическая анемия, а предваряющим для этого патологического состояния был хронический лимфолейкоз, назначают глюкокортикоиды как основной курс терапии. Эти препараты помогают при хроническом сублейкемическом течении лимфолейкоза, сопровождающемся сильным разрастанием печени, селезенки, лимфатических узлов. Глюкокортикоиды используют, если пациент не переносит цитостатические медикаменты, нет возможно применить облучение либо патологическое состояние проявляет стойкость к таким терапевтическим подходам.
Важные нюансы
Гормональные средства незаменимы, если цитостатические стали причиной цитопении, геморрагического синдрома. Их применяют в рамках полихимиотерапии, комбинируя основной курс и преднизолон.
Для описываемого патологического состояния характерны осложнения инфекционной природы. При таком развитии ситуации больному показан курс антибиотиков. Чаще всего применяют препараты обширного спектра эффективности. Хорошо зарекомендовали себя макролиды, аминогликозиды. Можно использовать полусинтетические средства из пенициллинового ряда, цефалоспоринового, иммуноглобулин.
Миелопролиферативный синдром
Это патологическое состояние нередко рассматривают в рамках образовательной программы вместе с описанным выше. Термином принято обозначать патологию, при которой активно вырабатываются миелоидные клетки. Причина явления – неправильная работа стволовых клеток системы, ответственной за производство крови. Синдром объединяет в себя несколько болезней – лейкоз, миелофиброз, тромбоцитоз, полицитемию. Сюда же принято относить миелодиспластический синдром.
Иммунопролиферативные заболевания условно разделяются на три категории:
- лимфопролиферативные нарушения. Классифицируются в зависимости от производства лимфоцитов (избыточного), то есть белых клеток крови. К ним относятся такие болезни, как множественная миелома, различные формы лейкемии;
- гипергаммаглобулинемия. Превышение нормального уровня иммуноглобулина в сыворотке крови;
- парапротеинемия. Наличие в крови повышенного уровня парапротеинов. Многие виды лейкемии и лимфом часто вызывают парапротеинемию.
Такие заболевания могут быть обнаружены и подтверждены при помощи МРТ, КТ, которые показывают точное расположение и размеры опухолей. Кроме того, широко применяется исследование образцов крови - биохимический анализ. Если определенные уровни клеток или химических веществ являются исключительно высокими (например, лейкоз вызывает аномально высокое количество лейкоцитов в крови), это может указывать на наличие серьезного заболевания. В зависимости от типа болезни пациенту может потребоваться пройти много стадий тестирования, прежде чем врач поставит окончательный диагноз. Дополнительные тесты могут включать взятие образцов костного мозга.
Лечение проводится при помощи химиотерапии, облучения, хирургического вмешательства или любой комбинации трех методов. Пациенты с поздней стадией лейкемии, например, часто подвергаются пересадке костного мозга для того, чтобы заменить раковые клетки здоровыми и повысить шансы на выживание.
Симптомы подобных нарушений варьируются, однако общими признаками иммунопролиферативных нарушений являются следующие состояния:
- боль в животе;
- анорексия;
- потеря веса;
- избыточная масса в животе (ощущение лишнего веса, вздутие, отечность);
- диарея и различные расстройства стула.
Кроме того, в эту общую группу входят такие нарушения, как:
- макроглобулинемия Вальденстрема;
- болезнь альфа-тяжелых цепей;
- болезнь гамма-тяжелых цепей;
- иммунопролиферативная болезнь тонкого кишечника.
Далее более подробная информация о каждом из этих состояний.
Макроглобулинемия Вальденстрема
Макроглобулинемия Вальденстрема - это одно из злокачественных моноклональных расстройств, относящихся к хроническим пассивным расстройствам. Оно характеризуется наличием высокого уровня макроглобулина (иммуноглобулин М или IgM), повышенной вязкостью сыворотки крови, а также наличием лимфоплазмоцитарных нарушений в костном мозге. Классическими проявлениями макроглобулинемии Вальденстрема являются:
- присутствие парапротеина IgM и злокачественного клеточного инфильтрата в костном мозге и других тканях тела.
Нередко это заболевание проявляется в виде макроглобулинемии и множественной миеломы, заболеваниях почек и литических изменениях костей.
- синдром повышенной вязкости крови (проявляется в кровотечениях, головокружении, головной боли, нечеткости зрения, проблемах со слухом);
- диарея;
- заболевания почек;
- амилоидоз сердца, почек, печени, легких, суставов;
- кровотечения как проявление дисфункции тромбоцитов и аномалии фибриногена;
- криоглобулинемия;
- повышенная предрасположенность к инфекциям из-за дисфункции В и Т клеток лимфоцитов;
- сердечная недостаточность;
- высокая частота появления лимфом, миелодисплазий и лейкозов.
Симптомы. Начало болезни коварное и неспецифическое. Многие пациенты живут с этим расстройством до тех пор, пока оно не обнаруживается при обычном анализе крови. Слабость, потеря аппетита и потеря веса так же характерны для макроглобулинемии Вальденстрема. Дополнительные симптомы: изменения психического состояния, в том числе вялость, оцепенение, или даже коматозное состояние, инфильтрация ЦНС (нейролейкемия), спутанность сознания, потеря памяти, дезориентация, моторные нарушения (синдром Бин-Нееля). Возможны визуальные отклонения, такие как затуманенное зрение или двойные изображения.
Болезнь альфа-тяжелых цепей
Болезнь альфа-тяжелых цепей - тип генетического нарушения, характеризующийся неполным циклом производства моноклональных альфа-тяжелых цепей без легких цепей. Считается подтипом иммунопролиферативного заболевания кишечника. Клиническая картина: хронический понос с признаками нарушения всасывания жидкости в кишечнике.
Распространенность заболевания неизвестна, но большинство случаев были зарегистрированы в Северной Африке, на Ближнем Востоке и в странах Средиземноморья, и были связаны с плохой санитарной обстановкой. В мировой медицинской отчетности зарегистрировано более 400 случаев заболевания.
Болезнь альфа-тяжелых цепей поражает пациентов молодых возрастных групп (от 20 до 30 лет). Все пациенты имеют признаки мальабсорбции, а также диарею, потерю веса и боли в животе. Часто присутствуют паразитарные инфекции, наблюдается инфильтрация слизистой оболочки тощей кишки с патологическими плазмоцитами клеток. По мере прогрессирования заболевания развивается иммунобластная лимфома. Лимфоплазматическая инфильтрация слизистой оболочки кишечника выражается как реакция пищеварительного тракта и иммунной системы на длительное антигенное воздействие кишечных организмов. Диагноз ставится на основании наличия свободных тяжелых альфа-цепей при отсутствии легких альфа-цепей. Структура таких цепей обычно укорочена. Эти альфа-цепи обнаруживаются в биологических жидкостях (сыворотка крови, моча, выделения тощей кишки и другие жидкости) при процедуре иммуноэлектрофореза, иммуноселекции или иммунофиксации.
Начальное лечение заключается в искоренении любой сопутствующей инфекции (например, паразиты, вирусы, Helicobacter Pylori, Campylobacter jejuni) соответствующими антибиотиками. Для пациентов с симптоматическим заболеванием, неадекватно реагирующим на антибиотики, рекомендуется химиотерапия по схеме лечения неходжкинской лимфомы. В случаях присутствия громоздких опухолей рекомендуется хирургическая резекция. Без антибиотиков и химиотерапии болезнь быстро прогрессирует и имеет в целом негативный прогноз.
Болезнь гамма-тяжелых цепей
Болезнь гамма-тяжелых цепей - это состояние, которое характеризуется производством неполных моноклональных гамма-тяжелых цепей без легких цепей, которые служат связующими звеньями цепи. Клиническая картина обычно напоминает системные лимфопролиферативные/аутоиммунные заболевания. Всего зарегистрировано порядка 120 случаев этого заболевания к настоящему моменту. Болезнь возникает у возрастной группы от 40 до 90 лет, однако изредка встречалась и у молодых людей. Рассеянное лимфопролиферативное заболевание с лимфаденопатией, спленомегалией и гепатомегалией является наиболее распространенным признаком болезни гамма-тяжелых цепей. Проявляется болезнь обычно в поражениях кожи и костного мозга. У некоторых пациентов может проявляться как ревматоидный артрит, аутоиммунная гемолитическая анемия или тромбоцитопеническая пурпура.
Поскольку болезнь гамма-тяжелых цепей является гетерогенным состоянием, выбор терапии должен основываться на симптомах основного заболевания. Любое аутоиммунное заболевание должно корректироваться при помощи стандартной терапии, с учетом аномального моноклонального компонента. Препараты для лечения болезни гамма-тяжелых цепей: Мелфалан, Хлорамбцил, Циклофосфамид и Винкристин. В некоторых случаях целесообразно использование лучевой терапии или хирургическое удаление экстрамедуллряных плазмоцитом (новообразований). Состояние характеризуется переменной клинической картиной и может быть как бессимптомным, так и переходящим к быстро прогрессирующей опухоли с плохим прогнозом.
Иммунопролиферативная болезнь тонкого кишечника
Иммунопролиферативная болезнь тонкого кишечника (IPSID) встречается в основном у молодых людей с низким социально-экономическим статусом, проживающих в развивающихся странах. Лимфоплазмоциты проникают в эпителий двенадцатиперстной и проксимальной тощей кишки, локализуются также в брыжеечных лимфатических узлах. В двух третях случаев привлеченные лимфоциты производят аномальные альфа-тяжелые белковые цепочки. Этиология этого заболевания не ясна, хотя учеными были предложены различные паразитарные, генетические и токсичные механизмы заражения.
Половина пациентов с этим состоянием была госпитализирована уже с наличием кишечной В-клеточной лимфомы, а у остальных лимфома развилась в течение нескольких лет после начала заболевания. Симптомами иммунопролиферативной болезни тонкого кишечника являются следующие состояния: расстройство стула, диарея, нарушение всасывания жидкости в кишечнике, тошнота, изжога, рвота, нарушение перевариваемости и усваиваемости пищи, и как следствие, похудение, быстрая потеря веса. При наличии вторичной инфекции лечение проводят сначала антибиотиками, а затем препаратами химиотерапии.
По материалам:
Pr Robert KYLE
1994-2015 by WebMD LLC: Karen Seiter, MD, Doris Ponce,
MD, Emmanuel C Besa, MD
The American Journal of Gastroenterology
2015 The American College of Gastroenterology

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Патогенез
- Симптомы
- Формы
- Диагностика
- Что нужно обследовать?
- Какие анализы необходимы?
- Дифференциальная диагностика
- Лечение
- Прогноз

Аутоиммунный лимфопролиферативный синдром (АЛПС) - заболевание, в основе развития которого лежат врожденные дефекты Fas-опосредоаанного апоптоза. Оно было описано в 1995 г., но еще с 60-х годов заболевание со схожим фенотипом было известно под названием синдром CanaLe-Smith.
Заболевание характеризуется хронической незлокачественной лимфопролиферацией и гипергаммаглобулинемией, которые могут сочетаться с различными аутоиммунными нарушениями.

[1], [2], [3], [4], [5]
Код по МКБ-10
Патогенез
Апоптоз, или физиологическая гибель клетки, является одним из неотъемлемых механизмов поддержания гомеостаза организма. Апоптоз развивается вследствие активации различных сигнальных механизмов. Особую роль в регуляции системы гемопоэза и иммунной системы играет апоптоз, опосредованный активацией Fas-рецепторов (CD95) при их взаимодействии с соответствующим лигандом (Fas-лиганд, FasL). Fas представлен на различных гемопоэтических клетках, высокая экспрессия Fas рецептора характерна для активированных лимфоцитов. Fasl-экспрессируется, главным образом, CD8+T-лимфоцитами.
Активация Fas рецептора влечет за собой ряд последовательных внутриклеточных процессов, итогом которых является дезорганизация ядра клетки, денатурация ДНК, изменения мембраны клетки, приводящие к ее распаду на ряд фрагментов без выброса во внеклеточную среду лизосомальных ферментов и без индукции воспаления. В передаче апоптотического сигнала к ядру участвует ряд ферментов, называемых каспазами, в том числе каспаза 8 и каспаза 10.
Fas-опосредованный апоптоз играет важную роль в элиминации клеток с соматическими мутациями, аутореамтивных лимфоцитов, а также лимфоцитов, выполнивших свою роль в процессе нормального иммунного ответа. Нарушение апоптоза Т-лимфоцитов приводит к экспансии активированных Т-клеток, а также так называемых двойных негативных Т-лимфоцитов, которые экспрессируют Т-клеточный рецептор с a/b цепями (TCRa/b), но не имеют ни CD4, ни CD8 молекул. Дефект программируемой гибели В-клеток в совокупности с повышением уровня интерлейкина 10 (IL-10) приводят к гипергаммаглобулинемии и повышению выживаемости аутореактивных В-лимфоцитов. Клиническими последствиями являются избыточное накопление лимфоцитов в крови и лимфоидных органах, увеличение риска аутоиммунных реакций и опухолевого роста.
К настоящему времени выявлено несколько молекулярных дефектов, приводящих к нарушению апоптоза и развитию АЛЛС. Это мутации в генах Fas, FasL, каспазы 8 и каспаэы 10.
[6], [7], [8], [9], [10], [11], [12], [13]
Симптомы аутоиммунного лимфопролиферативного синдрома
АЛПС отличается большой вариабельностью спектра клинических проявлений и тяжести течения, и возраст клинической манифестации также может колебаться в зависимости от выраженности симптоматики. Известны случаи дебюта аутоиммунных проявлений во взрослом возрасте, когда и был диагностирован АЛПС. Проявления лимфопролиферативного синдрома присутствуют с рождения в виде увеличения всех групп лимфоузлов (периферических, внутригрудных, внутрибрюшных), увеличения размеров селезенки, а часто и печени. Размеры лимфоидных органов могут изменяться в течение жизни, иногда отмечено их нарастание при интеркуррентных инфекциях. Лимфоузлы имеют обычную консистенцию, иногда плотноваты; безболезненны. Известны случаи резко выраженных проявлений гиперпластического синдрома, имитирующих лимфому, с увеличением периферических лимфоузлов, приводящим к деформации шеи, гиперплазией внутригрудных лимфоузлов вплоть до развития синдрома сдавления и дыхательной недостаточности. Описаны лимфоидные инфильтраты в легких. Однако во многих случаях проявления гиперпластического синдрома не столь драматичны, и они остаются незамеченными врачами и родителями. Степень выраженности спленомегалии также весьма вариабельна.
Тяжесть течения заболевания определяется главным образом аутоиммунными проявлениями, которые могут развиться в любом возрасте. Чаще всего встречаются различные иммунные гемопатии - нейтропения, тромбоцитопения, гемолитическая анемия, которые могут сочетаться в виде двух- и трехростковых цитопений. Может иметь место единичный эпизод иммунной цитопении, но зачастую они носят хронический или рецидивирующий характер.
Из других, более редких аутоиммунных проявлений, могут наблюдаться аутоиммунный гепатит, артрит, сиаладенит, воспалительные заболевания кишечника, узловатая эритема, панникулит, увеит, синдром Guiltain-Barre. Кроме того, могут наблюдаться различные кожные сыпи, преимущественно уртикарные, субфебрилитет или лихорадка без связи с инфекционным процессом.
У больных аутоиммунным лимфопролиферативным синдромом увеличена частота развития злокачественных опухолей по сравнению с популяцией. Описаны случаи гемобластозов, лимфом и солидных опухолей (карцинома печени, желудка).
[14]
Формы
В 1999 г. предложена рабочая классификация аутоиммунного лимфопролиферативного синдрома, основанная на типе дефекта апоптоза:
- ALP5 0 - полный дефицит CD95, являющийся следствием гомозиготной нуль-мутации (homozygous nuLl mutation) в гене Fas/CD95;
- ALPS I - дефект передачи сигнала через Fas-рецептор.
- При этом ALPS la является следствием дефекта Fas-рецептора (гетерозиготная мутация в гене Fas);
- ALPS lb является следствием дефекта Fas-лиганда (FasL), связанного с мутацией в соответствующем гене - FASLG/CD178;
- ALPS Ic является следствием только что выявленной гомозиготной мутации в гене FA5LG/CD178;
- ALPS II - дефект внутриклеточной передачи сигнала (мутация в гене каспазы 10 - ALPS IIа, в гене каспазы 8 - ALPS IIb);
- ALPS III - молекулярный дефект не установлен.
АЛПС 0 типа - полный дефицит CD95 - описан всего лишь у нескольких пациентов, Поскольку гетерозиготные члены семей не имеют фенотипа АЛПС, была предложена гипотеза о аутосомно-рецессивном типе наследования. Однако, неопубликованные данные о наблюдении за семьей, в которой был выявлен пациент с АЛПС 0, не полностью согласуются с данным утверждением. Ученые выяснили, что многие, если не все, мутации являются доминантными, и что если они оказываются гомозиготными, это приводит к более выраженному фенотипу заболевания.
При АЛПС I типа тип наследования - аутосомно-доминантный, с неполной пенетрантностью и вариабельной экспрессивностью. В частности, при АЛПС1а описаны случаи гомозиготности или сочетанной гетерозиготности, при которой определяются различные мутации гена Fas в обеих аллелях. Эти случаи характеризовались тяжелым течением с пренатальной или неонатальной манифестацией (водянка плода, гепатоспленомегалия, анемия, тромбоцитопения). Кроме того, выявлена корреляция выраженности клинической симптоматики с типом мутации в гене Fas; для мутации во внутриклеточном домене характерно более тяжелое течение. Всего в мире описано более 70 пациентов с ALPS la. Мутация FasL была впервые описана у пациента с клиническими проявлениями системной красной волчанки и хронической лимфопролиферацией. Она была категоризирована как ALPS lb, хотя фенотип не полностью отвечал критериям классического аутоиммунного лимфопролиферативного синдрома (двойные негативные Т-клетки и спленомегалия отсутствовали). Первая гомозиготная мутация А247Е в гене FasL (внеклеточный домен) была описана недавно, в 2006 году, Del-Rey M et al. у пациента с нелетальным АЛПС, что говорит о важной роли терминального домена FasL C0OH во взаимодействии Fas/FasL. Авторы предлагают в действующую классификацию аутоиммунного лимфопролиферативного синдрома внести подгруппу ALPS Ic.
АЛПС II типа наследуется по аутосомно-рецессивному типу, и у многих пациентов с этим типом заболевания отмечалась типичная клиническая и иммунологическая АЛПС, включая нарушенный Fas-опосредованный апоптоз, в реализации которого задействованы как каспаза 8 (вовлечена на ранних этапах межклеточной передачи сигнала на уровне взаимодействий TCR и BCR), так и каспаза 10 (вовлечена в апоптотический каскад на уровне всех известных рецепторов, которые индуцируют апоптоз лимфоцитов).
Более чем у 30 пациентов была выявлена клиническая картина АЛПС средней степени выраженности, включавшая в себя гипергаммаглобулинемию и повышенный уровень двойных негативных Т-клеток в крови, причем активированные лимфоциты пациентов с АЛПС III типа (именно так был назван этот синдром) показывали нормальную активацию Fas-опосредованного пути in vitro, и никаких молекулярных дефектов найдено не было. Возможно, причиной заболевания служат нарушений других апоптотических путей, опосредованных, например, Trail-R, DR3 или DR6. Интересным кажется наблюдение R. Qementi о выявлении мутации N252S в гене перфорина (PRF1) у больного с АЛПС III типа, у которого наблюдалось существенное снижение НК-активности. При этом автор отмечает, что значительная разница между частотой обнаружения N252S у пациентов с АЛПС (2 из 25) и частотой ее выявления в группе контроля (1 из 330) заставляет предполагать ее связь с развитием АЛПС в итальянской популяции. С другой стороны, F. Rieux-Laucat отмечает, что данный вариант мутации PRF1 выявлялся им у 18% здоровых и у 10% больных АЛПС (неопубликованные данные). И, кроме того, наряду с полиморфизмом N252S, им найдена мутация гена Fas у пациента с АЛПС и его здорового отца, что, по мнению F.Rieux-Laucat, говорит о непатогенности гетерозиготной мутации N252S в гене перфорина, описанной несколько ранее R. Qementi у пациента с АЛПС (мутация Fas) и крупноклеточной В-лимфомой. Таким образом, вопрос о причинах возникновения АЛПС III типа на сегодняшний день остается открытым.
[15], [16], [17], [18]
Читайте также: