
Экспрессия генов при колоректальном раке

Успехи современной клинической онкологии неоспоримы. Все более сложные операции, новые препараты, эффективные методы обезболивания и устранения мучительных симптомов. Мы в нашем блоге достаточно рассказывали о том, как сегодня можно продлить и облегчить жизнь пациентам даже на последних стадиях болезни.
Но, тем не менее, тысячи онкологических больных во всем мире ежедневно узнают, что опухоль, которая вчера поддавалась определенному лечению – сегодня снова растет или дает метастазы. Врачи регулярно оказываются в тупике: все положенные лекарства и методы лечения перепробованы, и эффективных для данного пациента – не осталось.
Однако даже из этого тупика можно найти выход. С развитием генетики и молекулярной биологии в руках онкологов оказался новый способ изучить опухоль, чтобы найти в ней уязвимые места.
Для этого используют молекулярно-генетическое тестирование – определение особенностей ДНК раковых клеток. Метод сложный технически, дорогой, требует специфических знаний от врача.
Исследование занимает 3 недели, стоит от 250 до 670 т.р. В результате врач получает отчет в 30 страниц сложной информации, которой он еще должен уметь воспользоваться. Но пациентам, которые уже было перестали надеяться, это дает дополнительное время жизни.
Сегодня мы хотим рассказать о том, как делается молекулярно-генетическое тестирование, в каких случаях оно может помочь пациенту и какие знания дает врачу.
Мы все – мутанты, это норма. Но некоторые мутации приводят к раку
Для этого все соматические клетки (те, из которых состоит организм), кроме эритроцитов, беспрестанно делятся.
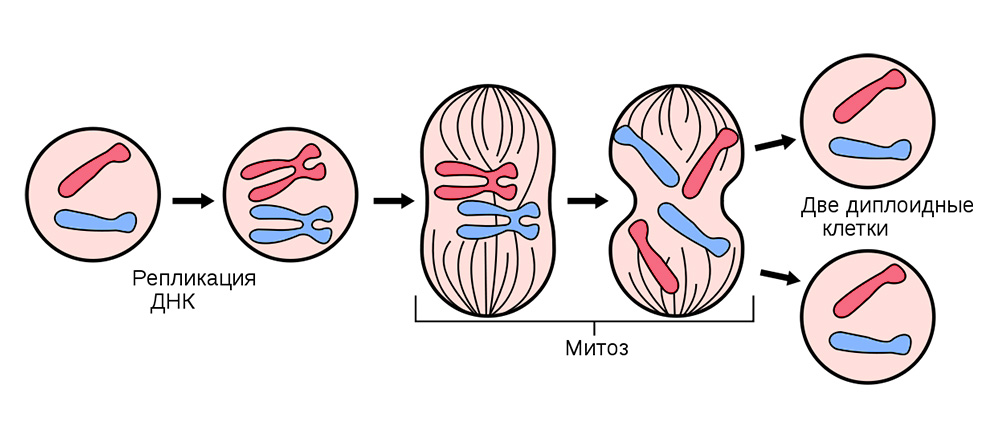
Деление соматических клеток происходит во всех органах и тканях
Иногда в процессе деления получаются сбои – мутации. То нить ДНК порвется, то скопируется с ошибкой, то участки хромосом перемешаются. Влиять на это может сотня факторов: от стресса и табачного дыма до воздействия радиации.
Мутации можно разделить на 4 вида.
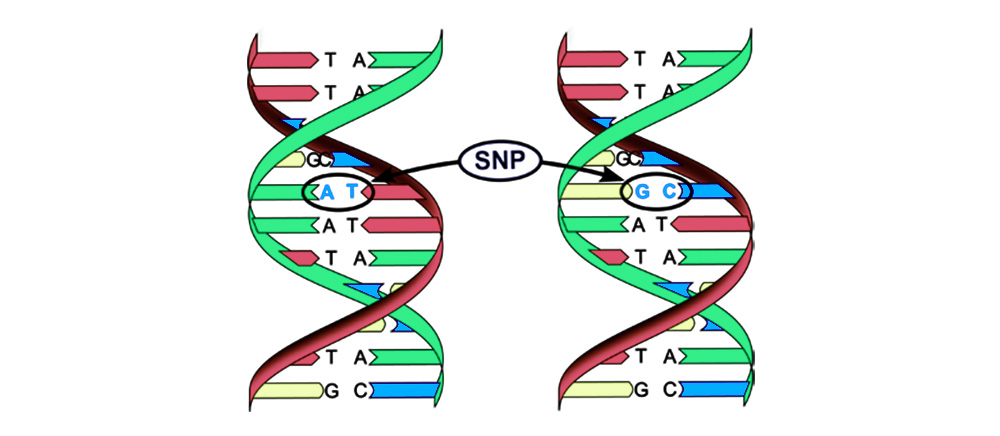
2. Хромосомные аберрации.
Делеция – утрата участка хромосомы. Происходят из-за обрыва концевого участка или разрыва ДНК сразу в двух местах. Всё – этот ген в хромосоме больше не экспрессируется.
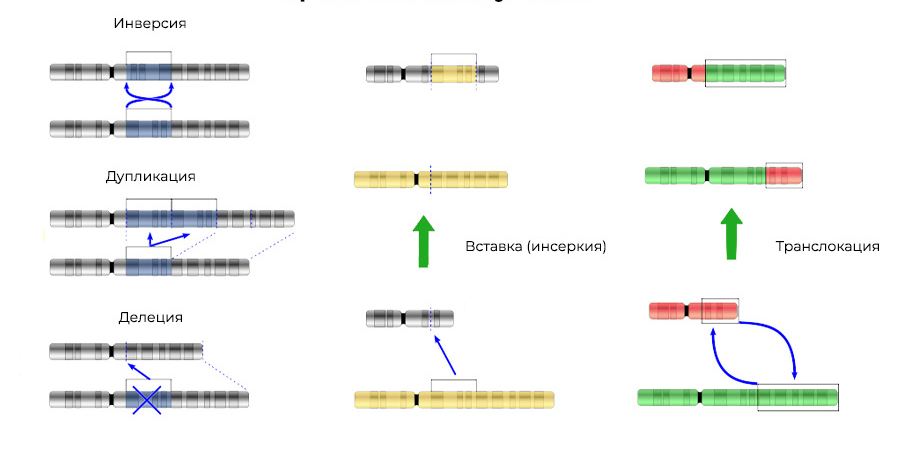
Мутации изменяют не только структуру участка ДНК, но и порядок этих участков
Злокачественную клетку от нормальной отличает нарушение клеточного цикла.
Клеточный цикл (жизнь клетки от деления до деления/гибели) строго регулируется работой специальных белков: киназы, циклины, факторы роста и транскрипционные факторы – в каждой живой клетке их десятки, и у каждого своя узкоспециальная, но важная функция.
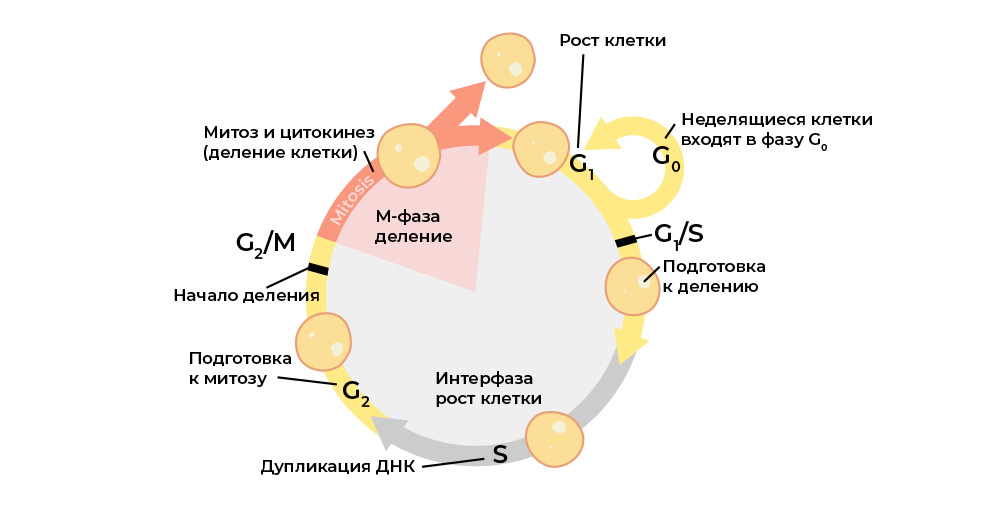
Каждый этап клеточного цикла контролируется белками-регуляторами
Таких значимых генов, изменения в которых могут привести к канцерогенезу (возникновению рака) – две больших группы.
Из тех, что наиболее хорошо изучены и у всех на слуху:
- EGFR, ALK, BRAF – немелкоклеточный рак легкого;
- BRAF – меланома;
- HER2 – рак молочной железы (РМЖ);
- KRAS – колоректальный рак.
Причем, мутации этих генов бывают обнаружены при нескольких видах опухолей. Например, повышенная экспрессия HER2 обнаруживается не только при РМЖ, но и при раке легкого и желудка.
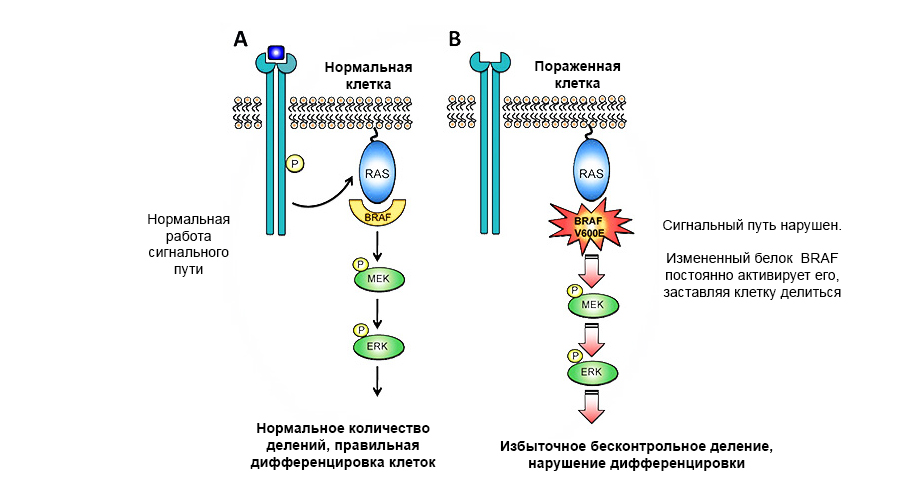
Мутация в протоонкогене белка BRAF приводит к неконтролируемому росту опухоли.
Гены-супрессоры опухоли (антионкогены) – напротив, могут подавить рост опухолевых клеток или участвуют в репарации (починке) поврежденной ДНК. А вот инактивация генов-супрессоров в результате мутации – резко увеличивает вероятность появления злокачественной опухоли.
Всего изучено влияние нескольких десятков протоонкогенов и опухолевых супрессоров на канцерогенез.
Зачем столько сложностей и как они продлевают жизнь пациентам
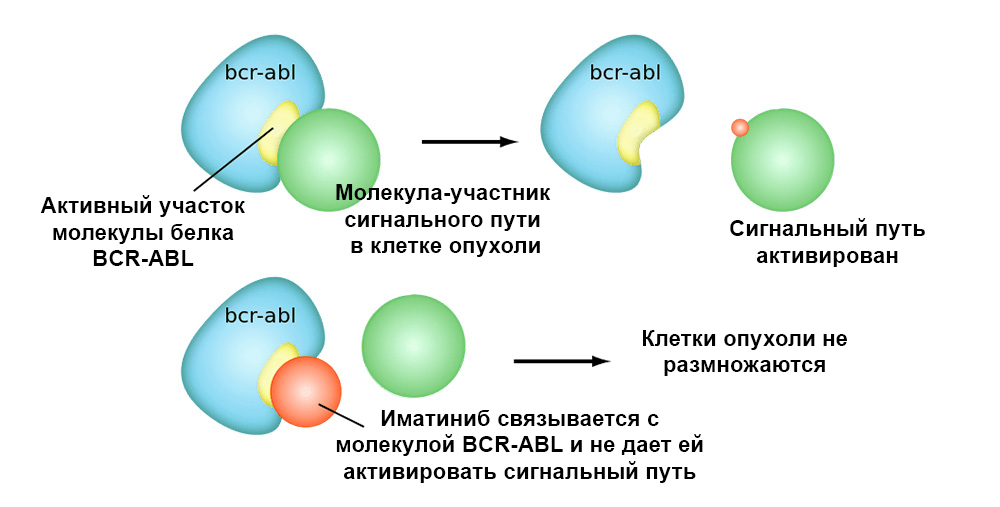
Иматиниб связывается с активным участком молекулы белка BCR-ABL, и блокирует его способность взаимодействовать с остальными молекулами в цепочках сигнальных путей.
Так что таргетные препараты имеют 2 важных преимущества перед классической химиотерапией.
Но и лечение для него должно быть соответствующее – индивидуально подобранное для конкретного пациента – на основе того, что мы определяем мутации в его опухолевых клетках.
В совсем недалеком прошлом злокачественные опухоли можно было классифицировать только по гистологии, то есть в зависимости от того, в каком органе они возникли, и как выглядели раковые клетки под микроскопом.
- узнаем чувствительность опухоли к препаратам;
- выясним, есть ли у опухоли устойчивость к определенным лекарствам;
- обнаружим генетические особенности, которые дают гиперчувствительность к препаратам;
- подберем новое лечение, если опухоль перестала отвечать на стандартную терапию;
обнаружим опухоль/метастаз на очень ранней стадии – по обрывкам ее ДНК в крови; - можем прогнозировать благоприятное или агрессивное течение заболевания.
Образцом выступает чаще всего ткань опухоли, либо взятая во время операции по удалению первичного очага, либо биопсия – микроскопический кусочек опухоли берут специальной тонкой длинной иглой.
Можно поискать ДНК опухолевых клеток в крови – тогда нужна так называемая жидкостная биопсия, две пробирки с кровью по 8,5 мл.
При биопсии мы часто сталкиваемся с тем, что многие пациенты боятся вообще трогать опухоль – опасаются, что ее это спровоцирует на рост. На сегодня не доступны какие-либо исследования, которые бы показали такую взаимосвязь. Конечно, биопсию надо выполнять правильно. У нас чаще всего врачи при заборе биоптата помечают место входа иглы: либо делают маленькую татуировочку (есть и такой инструмент ), либо скобку (хирургическую) ставят. Если потом понадобится операция, они иссекают весь этот ход, где была игла – от кожи до опухоли – так мы делаем шанс распространения раковых клеток за пределы опухоли еще меньше.
Далее образцы отправляются в лабораторию молекулярно-генетически исследований.
Причем применяют сразу несколько методов: секвенирования нового поколения (NGS), секвенирование по Сэнгеру и метод флуоресцентной гибридизации (FISH). Вместе они позволяют прочесть всю последовательность ДНК опухоли, выяснить драйверные мутации – то есть те, которые запустили злокачественный процесс и теперь могут быть мишенью для таргетной терапии – и даже визуализировать весь кариотип (хромосомный набор).
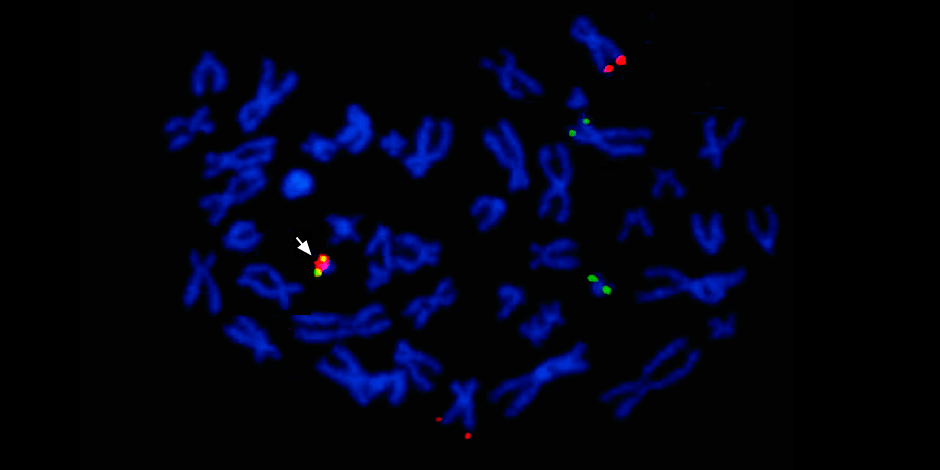
Под стрелкой слева – слияние красного и зеленого сигнала – свидетельство о слияния генетического материала хромосом 9 и 22 с образованием химерной филадельфийской хромосомы.
Кроме того, в полном молекулярно-генетическом исследовании обязательно определяют микросателлитную нестабильность (MSI, microsatellite instability) – нарушение в работе механизма репарации ДНК, которые приводят к быстрому накоплению мутаций в клетках. Этот фактор позволяет делать прогноз по поводу дальнейшего течения заболевания.
После получения молекулярно-генетического профиля опухоли – начинается его анализ
Специальные программы обрабатывают полученные результаты и составляют рекомендации автоматически. Но затем эти рекомендации обязательно вручную курируются командой экспертов. В анализе участвуют генетики, биоинформатики, врачи-онкологи, иммунологи и химиотерапевты. На этом этапе обязательно происходят уточнения и дополнения.
В итоге, в первой части отчета прописаны все найденные мутации в опухоли пациента, и таргетные препараты, которые будут наиболее эффективны в данном случае. Указана таргетная терапия, одобренная для данного типа опухолей с обнаруженными мутациями, и таргетная терапия, которая одобрена для лечения других типов рака с теми же мутациями. У нас в практике были случаи, когда назначались препараты именно второго порядка, off-label – и хорошо действовали.
Далее сотрудники лаборатории проводят огромную работу по мониторингу научных исследований, которые могут быть значимы в случае с данным пациентом.
Во второй части отчета находится обзор существующих на тот момент исследований с подробными данными о частоте встречаемости данной мутации, о действии разных препаратов и о возможности использовать тот или иной вид таргетной терапии при выявленных мутациях. Это помогает составить хотя бы приблизительный прогноз для пациента.
В третьей части отчета собраны актуальные клинические исследования, в которых пациент может принять участие, чтобы получить экспериментальное лечение. Это самый последний запасной способ, но знать о нем все подробности – полезно для спокойствия пациента.

Отчет получается довольно увесистым – 30 страниц захватывающего чтения
В этом случае молекулярно-генетическое исследование и дает нам понимание, какой препарат будет эффективен против данной опухоли, именно с этим набором мутаций. Назначение такого препарата позволяет выиграть главный для онкопациента ресурс – время.
Проблемы методики
Опухоли неоднородны. Они состоят из разных клеток, которые могут отличаться весьма значительно. И, например, в 80% клеток опухоли мутация определенного гена присутствует, а 20% клеток поделились с другим распределением хромосом – и остались немутировавшими. Да, мы назначаем препарат по результатам молекулярно-генетического теста, и против 80% опухолевых клеток он сработает эффективно, но для оставшихся 20% нужно будет придумывать другое лечение.
Некоторые виды рака более-менее гетерогенны, например, РМЖ. А некоторые опухоли, такие как саркомы, напоминают по структуре винегрет. Это затрудняет и диагностику, и лечение: нельзя заранее узнать, в какой части опухоли какие клетки, сколько их видов, как сильно они отличаются. И нельзя, грубо говоря, взять 10 образцов из разных мест опухоли – по ним придется сделать 10 отдельных генетических исследований.
До 30% таргетных и иммунопрепаратов в России назначается без соответствующего обоснования – без исследований генетики опухоли. И часть этих лекарств оказывается пустой тратой средств бюджета и денег пациента, потому что назначать таргетное лечение без понимания генетики опухоли – это рулетка: зарегистрировано более 600 препаратов. Например, для рака молочной железы есть пять протоколов лечения, в зависимости от мутации гена HER2/Neu.
В западной медицине определение генетического профиля опухоли уже становится стандартом лечения. Для российских онкопациентов молекулярно-генетические тестирования – все еще редкий случай, к сожалению – для бюджетной медицины это пока дорого. Но есть надежда, что все изменится к лучшему. Если сейчас оно стоит 600 тыс. руб., то 5 лет назад стоило больше миллиона – технология становится все проще и совершеннее, а, значит, популярнее и доступнее. Здесь время работает на нас.
Поэтому мало просо сделать генетический тест, нужно уметь понять результаты и сделать верные выводы. Мы с коллегами чаще всего сначала изучаем отчет сами (бывает, приходится посидеть над ним дома, в тишине после работы) – а потом еще и собираем консилиум, принимаем коллегиальное решение.
Но хорошие истории пациентов, честно говоря, всегда мотивируют лучше всего.
Сейчас у нас есть пациентка, 48 лет, с рецидивирующей глиобластомой (агрессивная опухоль мозга). К нам она попала после того, как прошла две линии терапии в государственном онкоцентре. Там все делали правильно, проводили лучевую терапию и назначали таргетный препарат, но опухоль все равно вернулась. Женщине отвели полгода жизни.
Мы предложили ей полное молекулярно-генетическое тестирование. Да, оно стоит 600 тыс. рублей, сокращенный вариант, за 250, в ее случае не подошел – нужно было расширенное тестирование, с максимально полным набором мутаций.
Но по результатам обследования назначили ей препарат, который предназначен обычно для лечения немелкоклеточного рака легкого. Он эффективен против опухолей с мутацией EGRF – у нашей пациентки глиобластома была именно с этой мутацией.
Женщина ходит к нам лечиться и наблюдаться уже 4 года. Это в 5 раз дольше, чем при стандартной терапии. Причем, она самостоятельна, живет эти 4 года обычной жизнью, ходит на работу и собирается дождаться внуков.
Предметы
Эта статья была обновлена
Аннотация
PROX1 является специфической мишенью пути β- катенина / TCF в кишечном эпителии. Он действует как регулятор прогрессирования от доброкачественного до высокодиспластического фенотипа при колоректальных опухолях. Однако клиническое значение экспрессии PROX1 не известно.
Методы:
Мы изучили прогностическую ценность иммуногистохимической экспрессии PROX1 в серии из 517 пациентов с колоректальным раком (CRC).
Результаты:
Большинство образцов опухолей экспрессировали PROX1 (91%, 471 из 517). Высокая экспрессия PROX1 была связана с плохой степенью дифференцировки опухоли ( P ![]()
Экспрессия PROX1 в образцах микрочипов ткани колоректального рака человека. ( A ) Низкая экспрессия PROX1 в ткани рака толстой кишки; только несколько ядер раковых клеток являются положительными. ( B ) Умеренная экспрессия PROX1 в ткани рака толстой кишки. ( C ) Сильная экспрессия PROX1 в ткани рака прямой кишки. ( D и E ) Очень сильная экспрессия PROX1 в ткани рака прямой кишки. ( F ) ткань рака прямой кишки, отрицательная для PROX1; однако смежные лимфатические эндотелиальные клетки (стрелка, вставка) окрашены положительно на PROX1. Шкала бар = 100 мкм .
Изображение в полном размере
Связь между экспрессией PROX1 и клинико-патологическими параметрами
Высокая экспрессия PROX1 была значительно чаще в опухолях высокой степени (3-4 степени) по сравнению с опухолями низкой степени (1-2 степени) ( P = 0, 0001; таблица 1). Ни одна из опухолей 1-й степени не имела высокой экспрессии PROX1. Не было обнаружено статистически значимой связи между PROX1 и возрастом на момент постановки диагноза, локализации опухоли, локализации опухоли, пола или стадии Герцога (таблица 1).
Ассоциация экспрессии PROX1 с CCSS
Для дальнейшего анализа мы разделили пациентов на две группы: низкий уровень PROX1 (баллы от 0 до 2) и высокий уровень PROX1 (баллы 3 и 4). Во всей серии пациентов высокая экспрессия PROX1 не была достоверно связана с CCSS (ОР = 1, 14; Р = 0, 38; Таблица 2, Рисунок 2А). 5-летний CCSS был 57% (95% доверительный интервал (ДИ), 52, 1–62, 5%) среди пациентов с низким уровнем экспрессии PROX1, и 53% (95% ДИ, 43, 6–62, 1%), когда экспрессия PROX1 была высокой. В подгруппе пациентов с раком толстой кишки высокая экспрессия PROX1 была связана с неблагоприятной выживаемостью (ОР = 1, 47; Р = 0, 045; Таблица 2, Рисунок 2B). 5-летний CCSS пациентов с раком толстой кишки с низкой экспрессией PROX1 составил 62% (95% ДИ, 55, 2–68, 9%), по сравнению с 47% для пациентов с высокой интенсивностью окрашивания (95% ДИ, 35, 3–59, 0%). 5-летний CCSS среди пациентов с раком толстой кишки с очень высокой (оценка 4; n = 11) экспрессией PROX1 составил всего 24%, что свидетельствует о том, что повышенная экспрессия опухолевых клеток PROX1 связана с худшим исходом у пациентов с раком толстой кишки. Кроме того, 5-летний CCSS женщин с раком толстой кишки с низкой экспрессией PROX1 (оценка 0–2) составил 63% (95% ДИ, 53, 3–73, 0%), по сравнению с 38%, когда экспрессия PROX1 была высокой (95% ДИ 22, 0–54, 7%; ОР = 2, 02; Р = 0, 007; фигура 2С), в то время как у мужчин с раком толстой кишки не было выявлено существенных различий (данные не представлены). Не выявлено значимой связи между PROX1 и выживаемостью у пациентов с раком прямой кишки. 5-летний CCSS для пациентов с низким уровнем PROX1 составил 51% (95% ДИ 43, 6–59, 4%), а у пациентов с высоким уровнем PROX1 - 61% (95% ДИ 47, 1–75, 9%; ОР = 0, 82, P = 0, 4; Таблица 2 Рисунок 2D).
Таблица в натуральную величину
![]()
Изображение в полном размере
Многомерный анализ выживаемости
Чтобы скорректировать установленные прогностические факторы при колоректальном раке, экспрессию PROX1 вводили в модель пропорциональных рисков Кокса вместе со стадией Дюкса, гистологической оценкой, возрастом на момент постановки диагноза, локализацией опухоли, локализацией опухоли и полом. В многомерном анализе выживаемости экспрессия PROX1 не была значимым прогностическим фактором (Таблица 3). Многофакторный анализ Кокса также был выполнен для подгруппы пациентов с раком толстой кишки. Однако экспрессия PROX1 не давала значимой прогностической информации в дополнение к выбранным факторам (данные не показаны).
Таблица в натуральную величину
обсуждение
Чтобы исследовать клиническую значимость PROX1, мы исследовали экспрессию PROX1 иммуногистохимией в образцах микроматрицы тканей 517 пациентов с CRC. Настоящие результаты показывают, что высокая экспрессия PROX1 связана с высокой степенью дифференцировки опухоли и менее благоприятным прогнозом в подгруппе пациентов с раком толстой кишки. Более того, наши данные показывают, что высокая экспрессия PROX1 связана с неблагоприятными исходами среди подгруппы пациентов с раком толстой кишки. Эти наблюдения согласуются с гипотезой о том, что избыточная экспрессия PROX1 способствует прогрессированию CRC (Petrova et al, 2008).
Мы обнаружили ядерную экспрессию PROX1 в 91% из 517 образцов CRC, и 27% этих образцов показали высокий уровень экспрессии. В настоящее время степень опухоли является важным клиническим показателем прогноза при CRC, и наше исследование показало, что высокая экспрессия PROX1 была связана с высокой степенью опухоли, но не с другими клинико-патологическими параметрами. Четкий уровень дифференцировки может указывать на различное биологическое поведение рака. В настоящем исследовании ни одна из опухолей 1-й степени не имела высокой экспрессии PROX1. Причина отсутствия экспрессии PROX1 в высокодифференцированных опухолях остается неизвестной, но это может быть связано с более низкой активацией пути Wnt. Для изучения механизма действия PROX1 необходимы дополнительные исследования.
Предыдущие исследования показали, что PROX1 действует как ядерный транскрипционный фактор (Oliver et al, 1993; Rodriquez-Niedenführ et al, 2001). На основании этих исследований и доклинических данных Petrova et al. (2008) мы решили оценить экспрессию PROX1 в соответствии с окрашиванием в ядрах опухолевых клеток. Однако также возможно, что PROX1 обогащается и / или активируется в цитоплазме перед транслокацией в ядро для выполнения его биологической функции, и, таким образом, окрашивание цитоплазматического PROX1 может быть детектируемым. Регуляция внутриклеточной локализации важна для действия транскрипционного фактора, и импорт ядер может служить механизмом регуляции экспрессии генов (rev. Yoneda, 2000). Кроме того, было показано, что Prospero, аналог Drosophila PROX1, часто обнаруживается в цитоплазме пролиферирующих и недифференцированных клеток (Li and Vaessin, 2000).
Текущее исследование показывает, что PROX1 имеет прогностическое значение среди пациентов с раком толстой кишки, тогда как у пациентов с раком прямой кишки различий обнаружено не было (Таблица 2, Рисунок 2). Среди пациентов с раком толстой кишки разница в выживаемости была очевидна у женщин, и высокая экспрессия PROX1 была связана с худшим исходом. Предполагается, что прогноз для правостороннего рака толстой кишки отличается от прогноза для левостороннего рака толстой кишки (rev. Distler and Holt, 1997). Различные причины такого различия могут включать факторы окружающей среды, генетические факторы и распределение по полу (обзор в Iacopetta, 2002). Во время эмбрионального развития правая кишка возникает из средней кишки, а левая - из задней кишки. Анализ генетических баз данных из нормальных образцов толстой кишки и опухолей выявил различия в экспрессии генов между нормальной слизистой оболочкой и карциномами толстой кишки, происходящими из правой и левой кишок (Glebov et al, 2003; Birkenkamp-Demtroder et al, 2005). Недавно в большом популяционном исследовании было показано, что правосторонний рак толстой кишки имеет худший прогноз, чем левосторонний рак, и что женщины с большей вероятностью получают правосторонний рак толстой кишки, хотя у женщин не было значительной разницы в смертности между рак левой и правой сторон толстой кишки (Meguin et al, 2008). В настоящем исследовании соотношение правосторонних и левосторонних опухолей было одинаковым у мужчин и женщин, и мы не обнаружили существенных различий в экспрессии PROX1 между правосторонними и левосторонними опухолями (данные не показаны).
По сравнению с другими типами рака, в CRC зарегистрировано очень мало молекулярных прогностических маркеров. Молекулярные маркеры опухолевой ткани были бы важны для принятия клинических решений, потому что целевая терапия - важная цель для улучшения результатов пациентов с CRC. На сегодняшний день точный механизм действия PROX1 в нормальной и больной ткани плохо изучен. Таким образом, необходимы дальнейшие исследования относительно молекулярных механизмов, которые регулируют экспрессию PROX1, и генов-мишеней прямой транскрипции PROX1. Таким образом, наши результаты показывают, что высокая экспрессия ядерного PROX1 связана с неблагоприятным исходом у пациентов с раком толстой кишки и, в частности, у женщин с раком толстой кишки. Кроме того, эти результаты подтверждают предыдущие доклинические наблюдения, предполагающие, что PROX1 играет роль в прогрессировании опухоли при CRC.
История изменений
Эта работа опубликована под стандартной лицензией на публикацию соглашения. Через 12 месяцев работа станет бесплатной, и условия лицензии перейдут на лицензию Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported.
Рак толстой кишки занимает 2-е место по смертности от злокачественных новообразований. По мере старения общества показатель летальности от рака этой локализации будет увеличиваться. Это неудивительно, если вспомнить об этиологии заболевания и особенностях патоморфологических изменений толстой кишки: ее слизистая оболочка представлена одноклеточным эпителием с быстрым периодом полного обновления и постоянной экспозицией к воздействию канцерогенных факторов.
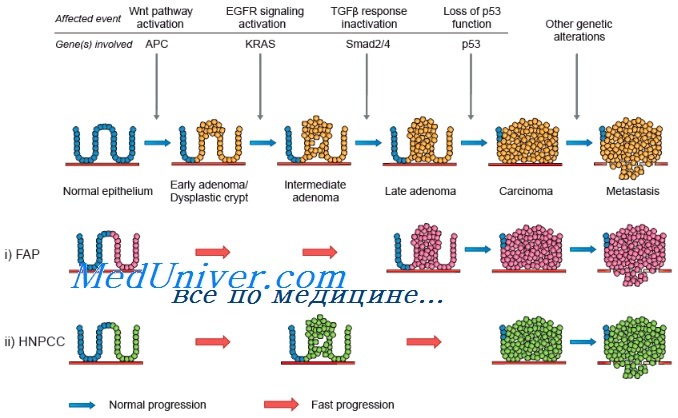
Следовательно, ДНК этих клеток постоянно и быстро делится и реплицируется. В результате вероятность накопления ошибок и злокачественной трансформации со временем увеличивается. Модель рака толстой кишки, разработанная Vogelstein и соавт., иллюстрирует эти механизмы. Мутации гена-супрессора аденоматозного полипоза толстой кишки (adenomatous polyposis coli, АРС) ведут к разрастанию полипов толстой кишки в раннем возрасте. Для уточнения характера любого заболевания толстой кишки можно легко получить образец ткани путем биопсии. По мере роста полипа он становится доступен для обследования гастроэнтерологом с использованием ряда исследований. Рак толстой кишки хорошо изучен также потому, что существуют семьи, члены которых подвержены редкому заболеванию — семейному аденоматозному полипозу толстой кишки. У этих больных толстая кишка покрывается сотнями и тысячами полипов, один или несколько из которых, как правило, малигнизируются в зрелом возрасте.
Именно у этой категории больных впервые был обнаружен ген АРС. В большинстве спорадических опухолей толстой кишки выявляют соматические мутации гена АРС. В более редких семейных случаях отмечается мутация гена АРС, передающаяся по наследству и в связи с этим присутствующая во всех эпителиальных клетках кишечника, что и предрасполагает к развитию полипов. Соматическая мутация ведет к активации онкогена ras и ускорению роста полипа, который, тем не менее, еще остается доброкачественным. Однако с годами полип может приобрести мутации генов-супрессоров DCC и ТР53, которые ведут к автономному неконтролируемому росту злокачественных клеток рака толстой кишки.
Семейная предрасположенность к раку толстой кишки достаточно широко распространена и делится на две группы: с образованием полипов и без них. Полипозы включают семейный аденоматозный полипоз толстой кишки, синдром Пейтца— Егерса, семейный ювенильный полипоз и гиперпластический полипоз. Риск рака толстой кишки у всех больных с названными синдромами повышен. Например, медиана возраста появления рака толстой кишки при семейном аденоматозном полипозе составляет 40 лет.
К синдромам без полипоза относится наследственный неполипозный колоректальный рак (ННПКРР). У этих пациентов злокачественную опухоль толстой или прямой кишки выявляют в возрасте около 40 лет. У большинства больных ННПКРР идентифицируют генетические дефекты в системе репарации ДНК. Мутации происходят в основном в гене MLH1, однако результаты последних исследований свидетельствуют о частом поражении таких генов, как MSH2 и MSH6. ННПКРР сопровождается и другими онкологическими заболеваниями: раком эндометрия (РЭ), раком яичника (РЯ), раком молочной железы (РМЖ) и опухолями нервной системы.
Семейный рак толстой кишки составляет примерно 3—5 % всех случаев рака этой локализации. Однако риск рака толстой кишки также повышен у родственников больных любой формой этого заболевания. Поэтому в большинстве случаев рака толстой кишки можно наблюдать, по крайней мере отчасти, наследственный компонент. Факторы окружающей среды играют важную роль в развитии заболевания; полученные данные указывают, что диета с высоким содержанием клетчатки может снижать вероятность развития рака толстой кишки. Фактор риска — рацион питания с высоким содержанием жира и красного мяса.
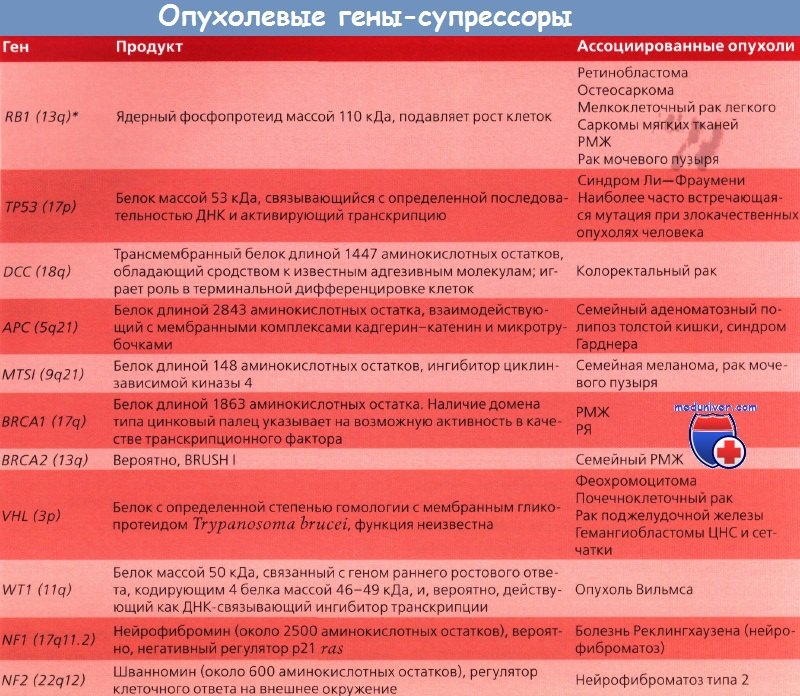
*Локализация на хромосоме
Молекулярные изменения, происходящие при раке толстой кишки, хорошо исследованы Vogelstein и соавт., подробно описавшими прогрессию нормального эпителия в рак. Первыми генами, для которых была установлена связь с раком толстой кишки, стали гены семейства ras, изначально выявленные в вирусах саркомы крыс. Эти гены наиболее часто обнаруживают в промежуточных полипах толстой кишки без дисплазии. В аденомах на ранних стадиях мутации ras встречаются редко, но в промежуточных полипах — регулярно, что свидетельствует о важной роли этих генов в раннем развитии рака толстой кишки.
Опухолевый ген-супрессор АРС принимает участие на самых первых этапах канцерогенеза при раке толстой кишки и был выявлен в исследованиях по изучению утраты гетерозиготности, показавших часто встречающуюся утрату длинного плеча хромосомы 5 (5q). Еще один ген, связанный с развитием рака толстой кишки, — ген-супрессор ТР53, расположенный на коротком плече хромосомы 17 (17р). Мутация этого гена — относительно позднее событие при раке толстой кишки. ТР53 — важный ген в контроле клеточного цикла и апоптоза. Утрата гена-супрессора DCC на длинном плече хромосомы 18 (18q) происходит на промежуточных и поздних этапах канцерогенеза при раке толстой кишки. Этот ген кодирует белок, ответственный за клеточную адгезию.
Ген DPC4, также расположенный на длинном плече хромосомы 18, вовлечен в сигнальную систему, связанную с трансформирующим фактором роста b.
При развитии рака толстой кишки происходит последовательное приобретение ряда мутаций. Для развития злокачественной опухоли обязательного прохождения всех этапов не требуется, однако предполагается, что необходимо по меньшей мере 6 или 7 генетических событий. У больных наследственным неполипозным колоректальным раком (ННПКРР) эти мутации развиваются вследствие неспособности эпителиальных клеток слизистой оболочки кишечника выявлять и устранять ошибки, происходящие во время нормального клеточного деления. Исследования Vogelstein и соавт. продемонстрировали модель, которая может быть применена и к другим опухолям.
Генетика семейного рака кишечника

Введение
Колоректальный рак (КРР) представляет собой заболевание со сложной этиологией. В его развитии важную роль играют диета и экологические факторы, в 15-30% случаев значимыми оказываются генетические факторы. Около 5% всех форм колоректального рака развивается на фоне хорошо известных наследственных синдромов, таких как синдром Линча (наследственный неполипозный колоректальный рак, ННКРР), семейный аденоматозный полипоз (САП) и MUTYH-ассоциированный полипоз (МАП).
О семейном колоректальном раке говорят в тех случаях, когда колоректальный рак встречается, но без доказательств в пользу наследственных синдромов. Риск развития колоректального рака увеличивается в 2-3 раза для субъектов, имеющих одного заболевшего КРР родственника первой линии родства, если на момент постановки диагноза возраст родственника превышал 50 лет. Для субъектов с двумя и более родственниками первой линии родства, заболевшими КРР в любом возрасте, или с одним родственником первой линии родства, заболевшим в возрасте до 50 лет, риск развития КРР увеличивается в 4-6 раз.
Для субъектов с умеренным риском развития КРР (RR >4) рекомендуется проведение колоноскопии каждые 3-5 лет, начиная за 5–10 лет до впервые установленного диагноза КРР у родственника или при достижении 45-летнего возраста [III,C].
Синдром Линча
Заболеваемость
Синдром Линча относится к заболеваниям, наследуемым по аутосомно-доминантному типу, представляя собой 3% всего КРР. Причиной его возникновения является мутация одного из генов, ответственных за ошибки репарации ДНК (mismatch repair ― MMR): MLH1, MSH2, MSH6 или PMS2.
Диагностика
Дефекты MMR сопровождаются неустойчивостью микросателлитов ДНК опухолевых клеток, что называется микросателлитной нестабильностью (МСН). При КРР, ассоциированном с синдромом Линча, эта молекулярная особенность обнаруживается в более чем в 90% случаев. Иммуногистохимическим методом, использующим антитела к четырем белкам ММR, может быть показана потеря экспрессии белка геном, вызывающим болезнь.
В настоящее время Амстердамские критерии II/ Исправленные критерии Bethesda (таблица №1) используются для отбора больных КРР, нуждающихся в исследовании микросателлитной нестабильности и/или иммуногистохимического анализа опухоли. А для пациентов с признаками наличия микросателлитной нестабильности или потери MMR экспрессии необходимо выполнение анализа мутации. В семьях с высокой вероятностью наличия мутации (Амстердамские критерии II, компьютерные модели) первостепенным методом является иммуногистохимический анализ, так как он может указать на необходимость выполнения анализа мутации. В других семьях на первом этапе можно использовать как исследование микросателлитной нестабильности, так и иммуногистохимический анализ.
При обнаружении потери экспрессии MLH1/PMS2 следует применять дополнительные анализы (BRAFV600E и анализ метилирования MLH1 промоутера) для исключение гиперметилирования MLH1 промоутера. Полученные результаты генеалогического анализа и MSI/IHC анализа необходимо обсуждать на мультидисциплинарных консилиумах (с участием патологоанатомов, клинических и молекулярных генетиков, гастроэнтерологов, хирургов, клинических онкологов и т.д.) [C].
Стадирование и оценка риска
Носители мутации гена MMR имеют высокий риск развития КРР (суммарный риск составляет 30-80%), рака эндометрия (суммарный риск составляет 3060%) и других ассоциированных опухолей (риск 30 с 10-99 аденомами или (2) один пациент в возрасте >30 с 10-99 аденомами и родственник первой линии родства, больной КРР с незначительным числом аденом. В обоих случаях в семье не должно быть родственников в возрасте моложе 30 лет с наличием более 100 аденом. У 25% пациентов из данной группы мутации могут обнаруживаться в АРС гене.
МАП. MUTYH-мутация обычно ассоциирована с вялотекущим полипозным фенотипом. Пациентов с более чем 10 полипами следует направлять на генетическую консультацию и проведение анализа на определение мутации MUTYH гена.
Стадирование и оценка риска
САП. Тяжесть полипоза толстой кишки коррелирует с локализацией мутации в АРС гене. У большинства пациентов сотни колоректальных полипов развиваются в детстве и юности. В возрасте до 10 лет КРР встречается крайне редко, а в возрасте 11-15 лет ― эпизодически. Без хирургического вмешательства к 40-50 годам у пациентов с САП почти неизбежно разовьется КРР. После выполнения колэктомии остается риск развития аденом прямой кишки. Существует риск возникновения аденом и даже рака в резервуаре после выполнения резекции прямой кишки. Аденомы также встречаются и в верхнем желудочно-кишечном тракте, особенно в двенадцатиперстной кишке. Если их оставить без лечения, то малигнизация развивается приблизительно в 5% случаев. Пациенты с САП также подвержены риску развития некоторых экстракишечных злокачественных и доброкачественных заболеваний. Рекомендации по наблюдению и лечению заболевания верхних отделов толстой кишки недавно широко обсуждались. Средний возраст пациентов с вялотекущим САП на момент диагностики КРР на 10-15 лет больше, чем при классическом САП. У пациентов с вялотекущим САП в отличие от пациентов с классическим САП может развиваться только несколько аденом в правой половине толстой кишки.
МАП. КРР, развивающийся в результате би-аллельных мутаций MUTYH, очень редко встречается в возрасте до 30 лет. У носителей би-аллельной мутации может развиваться небольшое количество аденом, а КРР чаще локализуется в проксимальной части толстой кишки. Аденомы (и рак) также развиваются в двенадцатиперстной кишке. Члены семьи с моно-аллельной мутацией MUTYH не находятся в группе риска развития КРР, поэтому они не нуждаются в регулярном выполнении колоноскопии.
Лечение
САП. Хирургическое лечение показано при большом количестве аденом более 5 мм, включая аденомы с высокой степенью дисплазии. Большинству пациентов с классическим САП хирургическое лечение выполняется в возрасте 1525 лет. К двум основным подходам в профилактической хирургии относятся резекция ободочной кишки с формированием илеоректального анастомоза (ИРА) и резекция ободочной кишки и прямой кишки с формированием резервуара из подвздошной кишки и илеоанального анастомоза (РИАА). Решение о выборе типа операции принимается в зависимости от многих факторов, включающих возраст больного, желания иметь детей, риска развития десмоида, локализации мутации в АРС-гене (при возможности) и тяжести ректального (и ободочного) полипоза. РИАА является предпочтительным лечением для пациентов с наличием большого количества ректальных аденом (>15–20 аденом). При наличии в прямой кишке небольшого количества аденом или при их отсутствии в прямой кишке возможно выполнение любого из двух лечебных подходов, что необходимо обсуждать с пациентом. Пациентам с множественными крупными (>5 мм) аденомами в прямой кишке, сопровождающимися высокой степенью дисплазии, после ИРА показана резекция прямой кишки.
МАП. Эндоскопическое удаление аденом возможно только в случае их небольшого количества. Если же необходимо выполнить операцию, то ИРА является достаточным в большинстве случаев. РИАА рекомендуется только в случае выраженного ректального полипоза.
NSAIDS (нестероидные противовоспалительные препараты) и/или специфические COX-2 ингибиторы уменьшают проявление колоректальных (и двенадцатиперстных) аденом. Хотя не известно предотвращают ли эти препараты риск развития КРР. В связи с тем, что недавно среди пациентов, получающих СОХ-2 ингибиторы, было описано появление сердечнососудистых побочных эффектов, использовать их следует только в крайних случаях.
Наблюдение при САП до операции
Классический САП: эндоскопическое обследование должно проводиться пожизненно. При бессимптомном носительстве мутации рекомендуется выполнение ректороманоскопии гибким фиброскопом каждые 2 года, начиная с 10-12-летнего возраста. При обнаружении хотя бы одной аденомы колоноскопию в последующем следует проводить ежегодно. Наблюдение среди субъектов высокого риска (родственники первой линии родства среди заболевших) в семье без обнаруженной АРС-мутации необходимо проводить с 2-х годичным интервалом до 40-летнего возраста, а последующие обследования до 50-летнего возраста можно выполнять с удлиненным интервалом (каждые 3-5 лет) [III, B].
Вялотекущий САП: рекомендуется выполнение колоноскопии каждые 2 года, начиная с 18-20-летнего возраста. При обнаружении хотя бы одной аденомы колоноскопию в последующем следует проводить ежегодно.
Наблюдение при МАП до операции
Обследование с выполнением колоноскопии должно проводиться пожизненно. При бессимптомном носительстве би-аллельной мутации рекомендуется регулярное обследование каждые 2 года, начиная с 18-20-летнего возраста [III, B].
Наблюдение при САП и МАП после операции
Наблюдение после ИРА: ректоскопию рекомендовано проводить с интервалом в 3-6 месяцев в зависимости от выраженности ректальных аденом.
Наблюдение после РИАА: Эндоскопическое исследование резервуара из подвздошной кишки рекомендовано проводить с интервалом в 6-12 месяцев.
Таблица №1. Амстердамские критерии (версия II, исправленная) и рекомендации Bethesda
Амстердамские критерии II: Должно быть, по крайней мере, 3 родственника с КРР или с опухолево-ассоциированным синдромом Линча: раком эндометрия, раком тонкой кишки, раком мочеточника или раком почки.
- один родственник должен быть родственником первой линии родства по отношению к двум другим;
- как минимум два последующих поколения должны быть поражены,
- по крайней мере, один случай обнаружения опухоли должен быть диагностирован до 50-летнего возраста,
- в любом случае возникновения КРР необходимо исключать САП.
Исправленные рекомендации Bethesda:
- КРР диагностируется у пациентов моложе 50 лет.
- Наличие синхронного, метахронного колоректального рака или опухолево-ассоциированного* синдрома Линча, независимо от возраста.
- КРР с фенотипом МСН в высокой степени, диагностированным у пациентов в возрасте до 60 лет.
- Пациент с КРР и родственником первой линии родства, страдающим синдромом Линча, ассоциированным с опухолью, диагностированной после 50 лет.
- Пациент с КРР с двумя или более родственниками первой или второй линии родства, страдающими синдромом Линча, ассоциированным с опухолью, независимо от возраста.
* опухоли, ассоциированные с синдромом Линча: колоректальный рак, рак эндометрия, рак желудка, рак яичников, рак поджелудочной железы, рак мочеточника, рак почки, опухоли билиарного тракта, опухоли головного мозга, аденомы сальных желез, кератоаконтомы, рак тонкой кишки.
Читайте также: