
Что такое примитивная нейроэктодермальная опухоль
Примитивная нейроэктодермальная опухоль (primitive neuroectodermal tumor — PNET) — новообразование детского возраста, в последние годы диагностируемое все чаще (1-10).
Ее классификация вызывает споры, в настоящее время на основании результатов сложных имму-ногистохимических и генетических исследований некоторые случаи экстраскелетной опухоли Юинга и других примитивных нейробластных опухолей были переклассифицированы в примитивные нейроэктодермальные опухоли.
Примитивная нейроэктодермальная опухоль развивается из клеток нервного гребня, возникает, как правило, у детей и характеризуется относительно низкой выживаемостью (1). Эта сложная проблема подробно изложена в соответствующих публикациях (1), в настоящем разделе мы излагаем лишь некоторые сведения о примитивной нейроэктодермальной опухоли глазницы (2-10).
а) Клиническая картина. Первичная примитивная нейроэктодермальная опухоль глазницы встречается в основном в детском и молодом взрослом возрасте. Обычно она сопровождается быстро развивающимся односторонним экзофтальмом и смещением глазного яблока. Она локализуется как в мышечном конусе, так и вне его (2-10), и вызывает изменения, аналогичные изменениям, наблюдаемым при рабдомиосаркоме.
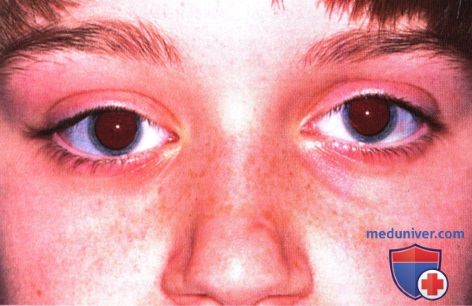
Примитивная нейроэктодермальная опухоль. Правосторонний экзофтальм у десятилетней девочки. 
КТ, корональная проекция: пациентка, представленная на рисунке выше; в верхневисочной части глазницы определяется опухоль, эрозия кости и гиперостоз. 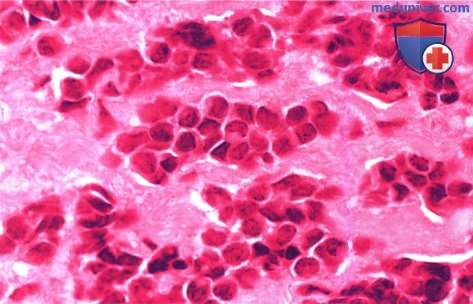
Гистологический препарат опухоли, показанной на рисунке выше: наблюдаются гнезда мелких клеток в строме фиброзной ткани (гематоксилин-эозин, х250).
б) Диагностика. По результатам лучевых исследований, опухоль может локализоваться в любой части глазницы. На ранних стадиях новообразование имеет четкие контуры, но вскоре становится инвазивным и часто вызывает деструкцию кости. При КТ и МРТ могут выявляться те же изменения, что и при метастатической нейробластоме глазницы, описанной ниже.
в) Патологическая анатомия. Примитивная нейроэктодермальная опухоль имеет несколько гистологических вариантов (1). Классическая опухоль состоит из пластов или долек, сформированных мелкими округлыми клетками, содержащими окрашивающиеся в темный цвет округлые или овальные ядра. Может присутствовать фиброзная соединительная ткань в различных количествах. Часто наблюдаются розетки Homer-Wright, иногда присутствуют розетки Flexner-Wintersteiner.
Как упоминалось выше, для уточнения диагноза необходимо проведение соответствующего иммуногистохимического исследования и консультация опытного патологоанатома.
г) Лечение. Показана биопсия, во время которой удаляется как можно больший объем ткани опухоли, после чего проводится химио- и, возможно, лучевая терапия; лечение осуществляется совместно с детским онкологом и лучевым терапевтом.
д) Список использованной литературы:
1. Smoll NR. Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer 2012;118:1313-1322.
2. Sen S, Kashyap S, Thanikachalam S, et al. Primary primitive neuroectodermal tumor of the orbit. J Pediatr Ophthalmol Strabismus 2002;39:242-244.
3. Alyahya GA, Heegaard S, Fledelius HC, et al. Primitive neuroectodermal tumor of the orbit in a 5-year-old girl with microphthalmia. Graefes Arch Clin Exp Ophthalmol 2000;238:801-806.
4. Kiratli H, Bilgic S, Gedikoglu G, et al. Primitive neuroectodermal tumor of the orbit in an adult. A case report and literature review. Ophthalmology 1999;106:98-102.
5. Bansal RK, Gupta A. Primitive neuroectodermal tumour of the orbit: a case report. Indian J Ophthalmol 1995;43:29-31.
6. Singh AD, Husson M, Shields CL, et al. Primitive neuroectodermal tumor of the orbit. Arch Ophthalmol 1994;112:217-221.
7. Wilson WB, Rolof F J, Wilson HL. Primary peripheral neuroepithelioma of the orbit with intracranial extension. Cancer 1988;62:2595-2601.
8. Chokthaweesak W, Annunziata CC, Alsheikh O, et al. Primitive neuroectodermal tumor of the orbit in adults: a case series. Ophthal Plast Reconstr Surg 2011;27: 173-179.
9. Romero R, Castano A, Abelairas J, et al. Peripheral primitive neuroectodermal tumour of the orbit. Br J Ophthalmol 2011 ;95:915—920.
10. Shuangshoti S, Menakanit W, Changwaivit W, et al. Primary intraorbital extraocular primitive neuroectodermal (neuroepithelial) tumour. Br J Ophthalmol 1986;70: 543-538.
Редактор: Искандер Милевски. Дата публикации: 23.5.2020


- Публикации
- Словарь терминов
- Мы и СМИ
- Новости




Примитивные или зрелые злокачественные нейроэктодермальные новообразования (neuroectodermal tumor) развиваются не из эпителия, как большинство раковых клеток, а из нейроэктодермы — клеточного материала головного или спинного мозга, вегетативной нервной системы и нейроэндокринных образований.
В зависимости от степени дифференциации и очага поражения нейроэктодермальные опухоли разделяются на две обширные группы: примитивные нейроэктодермальные опухоли кости (опухоли Аскина), являющиеся атипичным проявлением саркомы Юинга, и нейроэктодермальные опухоли — новообразования мягких тканей организма, чаще всего поражающие центральную нервную систему, а также головной мозг.
И в том, и в другом случае нейроэктодермальные опухоли относятся к опаснейшим онкологическим заболеваниям, которые чаще всего встречаются в возрасте от 3 до 20 лет, особенно у мальчиков.
Примитивная нейроэктодермальная опухоль фенотипически почти идентична саркоме Юинга. Это рыхлое мягкое злокачественное новообразование светло-серого цвета с обширными участками кровоизлияний и некрозов. Гистологические исследования характеризуют раковые клетки этой группы несколько меньшим содержанием гликогена, а также наличием в продуцированной ткани так называемых розеток Хомера-Райта, сформированных фибриллярным компонентом, протекающим между клетками. Также в опухолевом веществе часто встречаются кисты, возможно появление участков обызвествления и псевдоостеоидных образований — игл как при остеогенной саркоме хотя нейроэктодермальные иглы отличаются более упорядоченной структурой.
Опухоль состоит из незрелых мелких слабодифференциированных, быстро пролиферирующих раковых клеток, отличающихся высокой вирулентностью и способностью к инфильтрации. Наиболее типичные участки локализации опухоли Аскина — ребра, лопатки, грудная стенка, область малого таза, берцовые и бедренные кости. Разрушая кортикальный слой кости, опухоль быстро прорастает в окружающие ее мягкие ткани. Метастазы, в отличие от саркомы Юинга, способны распространяться не только в печени и других органах, но также проникать в сосуды и лимфатические узлы. Возможна инфильтрация в кости черепа, что в большинстве случаев приводит к летальному исходу. Если при саркоме Юинга пятилетняя выживаемость без возникновения рецидива болезни составляет порядка 70%, то при опухоли Аскина — не более 10%.
Среди нейроэктодермальных новообразований, поражающих мягкие ткани (ЦНС и оболочки мозга), выделяют внешне- и внутричерепные. Последние наиболее опасны, так как даже доброкачественные опухоли головного мозга обычно приводят к сдавливанию последнего стенками черепа или перекрытию ликворопроводящих путей, что способно вызвать тяжелейшую гипоксию и интоксикацию мозга. В нейроонкологии известны такие заболевания как астроцитома, глиобластома, эпендимома, медуллобластома, папиллома. Медуллобластома, особенно часто встречающаяся у детей (мальчиков 2-7 лет), — нейроэктодермальная опухоль мозга, образованная наиболее незрелыми клетками — медуллобластами. Продуцированные клетки медуллобластомы мелкие, слабодифференциированные, отличаются высочайшей вирулентностью и способностью к инфильтрации. В ткани опухоли встречаются розетки Хомера-Райта, кисты, а также детрит. Локализуются, как правило, в черве мозжечка, поражая ствол ЦНС, прорастая в мягкий кортикальный слой мозга и распространяясь метастазами по ликворопроводящим путям.
Симптомокомплекс при нейроэктодемальных опухолях характеризуется быстрым и болезненным при пальпации ростом новообразования. Если на начальном этапе развития саркомы боли носят местный и периодический характер, как правило, усиливаясь по ночам, то впоследствии усиливаются и становятся общими, существенно снижая качество жизни больного. Другими характерными симптомами являются лихорадка, тяжелая интоксикация, бессонница на фоне общей анемии, потеря аппетита, снижение массы тела, кашель, одышка. Поражение центральной или вегетативной нервной системы дополняет комплекс нейрологическими симптомами, среди которых повышенная возбудимость, раздражительность, страх.
Прогноз для любого типа нейроэктодермальных новообразований до сегодняшего дня остается крайне неблагоприятным, так как они очень плохо поддаются первичной диагностике. Большинство больных диагностируется чаще всего уже с обширными метастазами. При медуллобластоме мозга у детей прогноз относительно благоприятен, только если опухоль еще не успела диссеминировать. Отсутствие или незначительность метастазов гарантируют пятилетнюю выживаемость для 70% больных, хотя по статистике более чем у половины больных по истечении пятилетнего периода могут наблюдаться рецидивы. Для примитивной костной опухоли Аскина полное излечение на ранней стадии гарантировано в 10-20% случаев, пятилетняя выживаемость — в 62-70%.
Имеющиеся на сегодняшний день методы диагностики нейроэктодермальных опухолей включают рентгенографию, гистологические исследования, биопсию, внутривенное контрастирование, компьютерную и магнитно-резонансную томографию, радиоизотопное сканирование. Эти и другие методы позволяют максимально точно определить первичную локализацию опухоли, обнаружить метастазы в мягких костных тканях, а также в лимфатических сосудах и ликворопроводящих путях. Магнитно-резонансная томография дает возможность наиболее подробно изучить мягкие ткани, костный и головной мозг. С помощью МРТ нередко удается выявить самые мелкие очаги нейроэктодермальных новообразований, что особенно важно для благоприятного прогноза заболевания.
Лечение при нейроэктодермальных опухолях, как и при других онкологических заболеваниях, основано на наиболее рациональных схемах лучевой и полихимиотерапии. Хирургическое вмешательство показано в любом случае, хотя больше половины из них оказываются неоперабельными из-за обширной диссеминации. Считается, что примитивная нейроэктодермальная опухоль, будучи незрелой, плохо поддается воздействию лучевой терапии, однако облучение общей дозой не менее 20-60 Гр в сочетании с тщательно подобранной химиотерапией — единственно возможный способ лечения детей. При опухолях типа медуллобластомы производят интенсивное облучение мозжечкового червя, а также всего головного мозга и канала ЦНС. При костных новообразованиях воздействуют на пораженный участок. В химиотерапии применяют цитостатики, винкористин, актиномицин-Д, циклофосфамид, адриамицин.
Все применяющиеся на сегодняшний день методы лечения онкологических заболеваний даже если и способствуют выздоровлению, сопряжены с различными, в том числе тяжелыми, побочными эффектами, так как, убивая раковые клетки, лучевая и химиотерапия убивает и здоровые. Онкологи всего мира ищут способ, как сделать воздействие противораковой терапии точно направленным. До сего времени это им так и не удалось, однако недавно американскими учеными был найден вирус, который пролиферирует и паразитирует исключительно на измененных раковых клетках, не причиняя абсолютно никакого вреда клеткам здоровым. Неужели долгожданное, столь желанное для всего человечества, универсальное лекарство от рака наконец-то найдено?
Помочь детям с заболеванием нейроэктодермальные опухоли
На данный момент на попечении нашего фонда нет детей с данным диагнозом. Однако вы можете помочь больным детям с другими диагнозами!
Проанализированы собственные результаты лечения больных с первичной периферической примитивной нейроэктодермальной опухолью внутригрудной локализации. Наиболее убедительным подтверждением диагноза являются результаты морфологического и иммуногистохимического исследования опухоли. Отмечена эффективность проводимой химиотерапии во всех клинических наблюдениях, реализовавшаяся в частичной регрессии опухоли и разной степени выраженности лекарственного патоморфоза. Комбинированное лечение с неоадъювантной химиотерапией представляется наиболее оправданным и должно использоваться как метод выбора для данной категории пациентов.
Примитивная нейроэктодермальная опухоль (primitiv neuroectodermal tumor — PNET) входит в группу злокачественных опухолей, развивающихся из мигрирующих эмбриональных клеток неврального гребешка. Исследования последних десятилетий позволяют рассматривать данную опухоль в качестве одного из представителей группы близкородственных злокачественных новообразований, характеризующихся высокоагрессивным течением, а также наличием ряда тканеспецифических маркеров. Помимо PNET к данному семейству относятся саркома Юинга, в том числе экстраоссальная саркома Юинга. В 1979 г. американский морфолог F.B. Askin впервые выделил злокачественную
Особенностью морфологии саркомы Юинга и PNET является ее недостаточная патогномоничность для установления окончательного диагноза. Наибольшие сложности возникают при проведении дифференциальной диагностики с другими мягкотканными саркомами — рабдомиосаркомой, синовиальной саркомой, нейробластомой, лейомиосаркомой. Углубленное дифференциально-диагностическое обследование требует выполнения иммуногистохимического (ИГХ) анализа [3, 4]. В первую очередь следует исключить саркому Юинга, рабдомиосаркому, нейробластому. Для клеток саркомы Юинга и PNET характерна экспрессия виментина. В отличие от саркомы Юинга PNET характеризуется отчетливой нейроэктодермальной дифференцировкой, проявляющейся экспрессией нейроспецифической энолазы (NSE), синаптофизина, CD57, S-100, Leu-7 [3]. В первую очередь следует
исключить саркому Юинга, рабдомиосаркому, нейробластому. Опухоль Аскина характеризуется более выраженной митотической активностью [5].
Клиническая картина опухоли Аскина обусловлена наличием массивных опухолевых образований, распространяющихся в грудной полости и вовлекающих в патологический процесс органы средостения, плевру, легкие, грудную стенку. Как правило, на портяжении длительного времени заболевание протекает бессимптомно и лишь в поздней стадии манифестируется развитием плеврального выпота, проявлениями дыхательной недостаточности или кровохарканьем, симптомами интоксикации. Характерной особенностью патогенеза всех опухолей семейства PNET является ранняя инвазия в окружающие ткани и выраженная способность к гематогенному метастазированию. По данным А.И. Семеновой [3], все больные с локализованными формами этих опухолей имеют отдаленные микрометастазы.
Рентгенологическая диагностика, применяемая в полном объеме, имеет большое значение в выявлении опухоли, ее локализации, распространенности как первичного процесса, так и метастазов. Для уточнения протяженности изменений, точного стадирования опухоли и оценки динамики опухоли в процессе и после проводимого лечения необходимы КТ и МРТ-исследования с контрастированием [6]. При наличии у больного исходных или возникших на любом этапе лечения жалоб на боль в какойлибо точке костного скелета необходимо назначение остеосцинтиграфии, позволяющей диагностировать отдаленные метастазы в костях.
Результаты лечения в значительной мере зависят от распространенности опухоли. Большинство исследователей выделяют локализованные (операбельные) формы PNET и распространенные, обозначая их соответственно М0 и М1. Группировка по стадиям после операции предложена Национальным онкологическим институтом США. Стадия I — первичный очаг 5 см (нерезектабельная); стадия IV — определяются отдаленные метастазы. Рецидивы после удаления опухоли довольно характерны и чаще локализуются в зоне операции и в легких. Реже выявляют метастатические очаги в скелете, головном мозгу, печени [7].
Радикально удалить опухоль удается у 10–20%
больных [7, 8]. Средняя продолжительность жизни неоперабельных больных не превышает 8 мес [7]. Хотя многие авторы указывают на низкую чувствительность опухоли к химиотерапии (ХТ), у некоторых больных удается добиться частичной ремиссии или, по крайней мере, стабилизации [5, 7, 8].
ОБЪЕКТ И МЕТОДЫ ИССЛЕДОВАНИЯ
Мы располагаем наблюдением за 8 пациентами с первичной PNET грудной полости, находившимися на лечении в Национальном институте рака, за последние 5 лет. Возраст пациентов на начало лечения в клинике составлял от 2,5 до 22 лет. У 7 из них была односторонняя интраторакальная локализация опухоли с вовлечением от 2 до 6 ребер. У 1 пациента один мягкотканный опухолевый узел располагался в заднем средостении справа, второй опухолевый узел — в нижней доле левого легкого. Локализацию и распространенность опухолевого процесса во всех случаях определяли рентгенологическим обследованием, включая КТ органов грудной и брюшной полости (КТ ОГП, ОБП). По показаниям выполняли МРТ и остеосцинтиграфию. Во всех случаях диагноз был верифицирован до начала лечения морфологическим и ИГХ исследованием. Всем больным лечение было начато с назначения полихимиотерапии (ПХТ). Больному с опухолью Аскина (медиастинально-легочной локализацией патологического процесса) ПХТ проводили по схеме CWS-96, при PNET назначали лечение по схеме VIDE, пациенты получили от 3 до 6 циклов ПХТ. В дальнейшем все пациенты были прооперированы. Адъювантное лечение — по показаниям.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В результате неоадъювантного лечения у 1 пациента с PNET зарегистрирована полная рентгенологическая и морфологическая регрессия опухоли, у 7 — частичная регрессия (рис. 1). Хирургические операции после неоадьювантной ПХТ пациентам с PNET, выполненные в радикальном объеме, заключались в удалении внутригрудного компонента опухоли с резекцией от 2 до 6 ребер с последующей
пластикой грудной стенки. Из 7 пациентов с PNET безрецидивная выживаемость > 1 года после окончания лечения отмечена у 2 (28,6%), пролонгация заболевания до года выявлена у 2 (28,6%), 3 больных продолжают получать ПХТ в адъювантном режиме.
Рис. 1. Частичная регрессия PNET после неоадъювантной ПХТ. КТ пациента Б., 21 год: а, б — до лечения, в, г — после лечения
Приводим детальное описание наблюдения, представляющего интерес как с диагностической точки зрения, так и в отношении выбора метода лечения, а также его результатов. Анализ возникавших при этом проблем поможет в онкологических клиниках выработать оптимальную схему лечебных мероприятий в каждом индивидуальном случае.
г. ему была выполнена правосторонняя эксплоративная торакотомия, биопсия опухоли средостения. Гистологическое заключение: злокачественная низкодифференцированная опухоль (PNET).
При обращении в институт в июле 2007 г. предъявлял жалобы на боли в грудной полости, больше слева, общую слабость, потерю массы тела. Общее состояние пациента удовлетворительное, несколько снижен аппетит, периферические лимфоузлы не увеличены. На КТ ОГП слева мягкотканное узловое образование 98×61×82 мм, не отделяющееся от контура сердца, занимает нижнюю долю левого легкого, прорастает в средостение. Справа 2 узловых образования, одно — 71×39 мм — занимает средний этаж заднего средостения, проецируется на прикорневую зону нижней доли правого легкого, интимно прилежит к передним отделам сердца и, скорее всего, прорастает в перикард. Второе располагается ниже в заднем средостении, в заднем кардио-диафрагмальном углу (рис. 2). Учитывая имеющееся гистологическое заключение исследованного материала биопсии опухоли и данные КТ, установлен диагноз «примитивная нейроэктодермальная опухоль заднего средостения справа и нижней доли левого лег-
Рис. 2. КТ ОГП пациента С., 20 лет, до лечения: а — узловое образование в заднем средостении, б — узловое образование в нижней доле легкого
После проведенной терапии на контрольной КТ ОГП от 8.11.07 г. отмечена частичная регрессия опухоли в средостении. Рекомендовано провести еще 3 блока ПХТ по описанному выше протоколу. При контрольном обследовании 15.04.08 г. на КТ ОГП слева в прикорневой зоне нижней доли паравертебрально определяется патологическое образование 72×56×60 мм, с участками некроза в центре, четкими контурами. В заднем средостении справа опухолевая тень 37×26 мм распространяется на прикорневую зону нижней доли.
16.04.08 г. больной госпитализирован в отделение опухолей грудной полости института; 24.04.08 г. прооперирован. Опухоль в нижней доле левого легкого 70×60×60 мм, плотной консистенции, спаянная с перикардом, с пищеводом. Выполнена нижняя лобэктомия. Патогистологическое заключение (ПГЗ) № 10845-48/08 от 6.05.08 г.: злокачественная низкодифференцированная опухоль типа нейроэпителиомы (PNET) с массивными некрозами ткани опухоли, относительная жизнеспособность опухолевой ткани (ОЖОТ) — 73,1 ± 0,8%. На контрольной КТ ОГП и ОБП от 7.05.08 г.: слева — состояние после нижней лобэктомии. В заднем средостении справа патологическое образование размерами 34×21 мм, распространяющееся на прикорневую зону нижней доли (образование в прикорневой зоне 18×22 мм, в динамике от 6.09.07 г. уменьшилось в размерах). В легких очаговых образований не определяется. ОБП и забрюшинные лимфоузлы без патологии. 8.05.08 г. пациент выписан в удовлетворительном состоянии.
12.05.08 г. больной вновь госпитализирован для про-
должения лечения. 13.05.08 г. проведена операция — удаление опухоли заднего средостения справа с резек-
цией перикарда. Опухоль в заднем средостении с инвазивным ростом по задней стенке перикарда, размеры опухолевого узла около 2 см в диаметре, опухоль удалена с резекцией задней стенки перикарда. В корне легкого, между нижней и верхней легочными венами, опухоль около 2,5×2,5 см, опухолевый узел удален. ПГЗ № 11994-98/08 от 20.05.08 г.: низкодифференцированная злокачественная опухоль (PNET) с обширными некрозами, кровоизлияниями, фиброзом, периваскулярным склерозом. ОЖОТ — 2,5 ±0,9%.
При контрольном наблюдении за пациентом на протяжении 2008–2010 гг. (периодичность контрольных осмотров 1–3–6 мес) прогрессирования заболевания не определяли. Самочувствие хорошее, сохранял трудоспособность. 24.01.2011 г. обратился с жалобами на боли в левой стопе. Объективно — при пальпации отмечается болезненность свода левой стопы. На рентгенограммах костей стопы деструктивных изменений не выявлено. 28.02.11 г. на МРТ левой стопы определено неоднородное понижение структуры в медиальной клиновидной кости, за пределы кости процесс не распространяется. Заключение: больше данных о метастазе в медиальную клиновидную кость, исключить асептический некроз сложно. На фоне проведения противовоспалительной терапии боли в стопе усилились, появились боли в 1-м пальце левой стопы. На рентгенограмме костей стопы выявлена деструкция фаланги 1-го пальца. Заключение: метастазы PNET в кости левой стопы. 24.03.11 г. произведена трепанобиопсия клиновидной кости. ПГЗ № 1237 от 29.03 г.: метастаз PNET. ИГХ исследование №172/11 от 31.03.11 г. Маркеры: Anti-Synaptophysin Clone SY38 (+), Anti-Human CD99, Mic2 Gene Product (+), Anti-Vimentin Clone V9 (+). Заключение: метастаз PNET.
С 01.04.11 г. начата ПХТ по схеме VAID + бисфосфонаты в условиях стационара Национального института рака. Всего проведено 5 циклов ПХТ, последний цикл закончен 17.08.11 г. Между 3-м и 4-м циклами ПХТ проведен курс дистанционной телегамматерапии на метастазы в костях левой стопы по 2,5 Гр ежедневно, СОД 45 Гр. КТ ОГП, ОБП, костей левой стопы, свода черепа от 06.12.11 г. Заключение: состояние после комбинированного лечения PNET средостения и левого легкого. Данных о рецидиве опухолевого роста нет. Метастаз в клиновидной кости левой стопы, состояние после ПХТ+ЛТ, стабилизация процесса (деструкция ограничена зоной склероза). При контрольном осмотре в марте 2012 г. состояние больного вполне удовлетворительное, сохраняет трудоспособность. Таким образом, представленное наблюдение может заинтересовать со многих точек зрения. Пациенту в хирургическом отделении без уточненного генеза опухоли была выполнена операция, которая закончилась эксплоративной торакотомией с биопсией в связи с неоперабельным опухолевым процессом. Диагноз установлен при стандартном морфологическом исследовании. Данные КТ при первичном обращении указывают на двустороннюю локализацию опухолевого процесса в грудной полости, в заднем средостении и в лег-
ком, что уже представляет редкость для опухоли Аскина. В доступной литературе имеется лишь единичное сообщение Cevallaro Catalan R.I., Murphy Th. (1997) об изолированной первичной PNET легкого (цит. по [7]).
Первоначально опухолевая масса в средостении и в легком была больших размеров — признак неблагоприятного прогноза заболевания. Однако дальнейший процесс лечения и динамика заболевания сопровождались благоприятными прогностическими признаками. Так, в результате проведенной ЛТ и ПХТ получена частичная регрессия опухолевых узлов. Выполненные хирургические операции позволили радикально удалить остаточные опухолевые образования, локализующиеся в легком и средостении. Морфологическое исследование удаленных опухолей показало наличие терапевтического патоморфоза, особенно выраженного в опухоли средостения. При контрольном наблюдении за пациентом через 2 года 7 мес после окончания лечения констатировано появление симптомов метастазирования в кости стопы. Своевременно начатое комплексное лечение дало возможность локализовать дальнейшее распространение заболевания и стабилизировать метастатический процесс. Результат лечения — 5-летняя выживаемость пациента.
Высокоагрессивный характер PNET и саркомы Юинга в целом определяет неблагоприятный прогноз. Индивидуальный прогноз базируется на таких основных факторах как размер первичной опухоли, ее локализация, степень распространенности, эффект лекарственной терапии [9]. Важное прогностическое значение имеет длительность времени до прогрессирования. 5-летняя выживаемость больных, имеющих ранние, в течение первых 2 лет после проведенного лечения, рецидивы опухолевого процесса, не превышает 4,0–8,5%. Прогрессирование заболевания в более поздние сроки характеризируется показателями общей выживаемости 25–35% [10]. Фактором благоприятного прогноза является регресс опухоли в результате проведенной ХТ. По данным ряда авторов, некроз всей или большей части опухоли коррелирует с высокой, достигающей 85–95%, 5-летней выживаемостью больных [11].
Таким образом, PNET и опухоль Аскина — редкие злокачественные опухоли мягких тканей, встречающиеся чаще в молодом возрасте. PNET, которая локализуется в грудной полости, как правило, прорастает в грудную стенку с деструкцией костных структур. Наиболее убедительное подтверждение диагноза PNET, в частности опухоли Аскина, основывается на определении тканевых маркеров при ИГХ исследовании, одна-
ванием, можно утверждать, что комбинированное лечение с использованием неоадъювантной ХТ представляется наиболее оправданным.
Давыдов МИ, Мачаладзе ЗО, Полоцкий БЕ и др. Мезенхимальные опухоли средостения (обзор литературы). Сибирск онкол ж 2008; 1 (25): 64–74.
Алиев МД, Мехтиева НИ, Бохян БЮ. Факторы прогноза саркомы мягких тканей. Вопр онкол 2005; 46 (3): 47–52.
Семенова АИ. Саркома Юинга: характеристика заболевания, особенности диагностики, лечебная тактика. Практ онкол 2010; 11 (1): 45–52.
de Alava BE, Gerald WL. Molekular Biolodgy of the Ewings Sarcoma. Primitive Neuroectodermal Tumor Family. J Clin On- col 2000; 18 : 204–8.
Карташова ОМ, Анненкова ИВ, Карташов М.В. Лучевая диагностика периферической примитивной нейроэктодермальной опухоли у детей. Детская больница 2010; 3 : 47–52.
Neef H. The role of surgery in diagnosis and treatment of mediastinal malignance. Lung 2010; 168 , (Suppl 1): 1153–61.
Cotteril SJ, Abrens S, Paulussen M, et al. Prognostic factors in Ewings tumor of bone: Analisis of 975 patients from the Euro- pean Intergroup Cooperative Ewings SarcomaStudy Group. J Clin Oncol 2000; 18 : 3108–15.
Barker LM, Pendergrass TD, Sanders JE, et al. Survaival after recurrence of Ewings sarcoma family of tumors. J Clin On- col 2005; 23 : 4354–62.
Picci P, Bobling T, Bacci G, et al. Chemotherapy — induced tumor necrosis as a prognostic factor in localized Ewings sarcoma of the extremities. J Clin Oncol 1997; 15 : 1553–9.
Что такое саркома Юинга?
Саркома Юинга — это редкая опухоль кости, которая чаще всего встречается у подростков. Она может также возникнуть вне кости в мягких тканях. Саркома Юинга также связана с другим типом опухоли, известной как примитивная нейроэктодермальная опухоль (ПНЭО). Исследователи выяснили, что эти опухоли связаны с одной и той же хромосомной аномалией (сбалансированная взаимная транслокация) и имеют много физиологических характеристик. Следовательно, эти опухоли иногда совместно классифицируют как опухоли семейства Юинга (ОСЮ). Этот общий термин охватывает саркому Юинга кости, внекостной саркомы Юинга, примитивную нейроэктодермальную опухоль и опухоль Аскина (опухоль грудной стенки).
Саркома Юинга костей составляет около 70% опухолей в этом семействе. Как правило, термин саркома Юинга является предпочтительным, поскольку, несмотря на разные названия, молекулярно это одна опухоль. Саркома Юинга чаще всего поражает длинную кость ног (бедренную кость) и плоские кости, такие как кости таза и грудной клетки.
Саркома Юинга — это агрессивный рак, который может распространяться (метастазировать) в легкие, другие кости и костный мозг, потенциально вызывая опасные для жизни осложнения. Точная причина этих опухолей неизвестна.
Саркома Юинга была впервые описана в медицинской литературе в 1921 году доктором Джеймсом Юингом. Саркома Юинга является второй наиболее распространенной первичной опухолью костей у детей и составляет приблизительно 2% всех детских диагнозов рака.
Признаки и симптомы
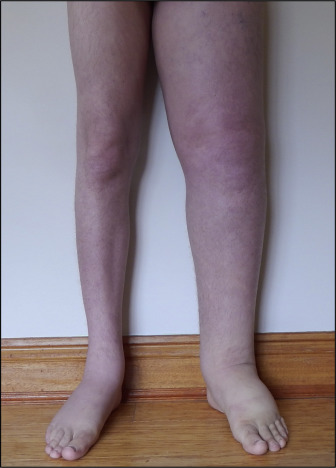
У людей с опухолью семейства Юинга могут проявляться боль и припухлость вблизи пораженной части тела. Боль часто приходит и уходит (перемежается) вначале, постепенно прогрессируя, становясь более последовательной. Слабость и онемение в зоне поражения также могут возникнуть. В некоторых случаях больные могут также испытывать лихорадку, недостаток энергии, потерю веса, низкий уровень циркулирующих эритроцитов (анемия) и повышенный уровень циркулирующих лейкоцитов (лейкоцитоз). Часто присутствует пальпируемая масса.
Саркома Юинга чаще всего поражает среднюю часть (диафизарную область) длинных костей рук и ног, особенно длинную кость голени (бедренная кость). Эти опухоли также обычно поражают плоские кости, такие как кости таза, грудной стенки и позвоночника (позвонков). Саркома Юинга может возникнуть в любой кости тела, например, кости стопы, кисти, нижней челюсти, черепа и/или других местах. Опухоли мягких тканей развиваются чаще всего в туловище и груди. Тем не менее, наиболее распространенным местом расположения является таз, составляющий около 25% случаев. Саркома Юинга может ослабить кости, иногда приводя к переломам.
Эти опухоли часто агрессивны и могут распространяться (метастазировать) на дополнительные участки тела, особенно на другие кости и легкие. В редких случаях может затрагивать костный мозг.
Симптомы, связанные с этими опухолями, являются вторичными по отношению к их расположению. Например, опухоль ноги может привести к хромоте, опухоль в легких может привести к проблемам с дыханием и скоплению жидкости в слоях ткани, которые выстилают легкие и грудную полость (плевральный выпот), или опухоль в позвоночнике может вызвать слабость или паралич пораженных мышц (параплегия).
Причины
Точная причина саркомы Юинга неизвестна, и основной тип клеток не был идентифицирован. Считается, что большинство случаев происходит случайно, без какой-либо конкретной причины (спорадически).
Причина, по которой происходит хромосомная транслокация между хромосомами 11 и 22, также неизвестна. Однако, по некоторым оценкам, более 85 процентов опухолей в семействе опухолей Юинга имеют эту транслокацию. Реже ген EWS может сливаться с другим геном, отличным от гена FLI; это часто гены из того же семейства, что и FLI1, чаще всего с участием гена ERG.
В очень редких случаях саркома Юинга может развиться как второе злокачественное новообразование, что означает, что расстройство развивается как позднее осложнение более раннего лечения другой формы рака.
Затронутые группы населения
Саркома Юинга поражает мужчин чаще, чем женщин. Заболевание может повлиять на людей любого возраста, но чаще всего встречается у людей 10-20 лет. Ежегодная заболеваемость составляет 2,93 ребенка на 1 000 000 человек. Приблизительно 200-250 детей и подростков ежегодно диагностируют опухоль семейства Юинга. Две трети выживают в течение длительного времени (более пяти лет). Опухоль встречается с большей частотой у европеоидной расы и чрезвычайно редко у афроамериканцев и азиатов.
Исследования показали, что между внекостной (экстраоссальной) саркомой Юинга (ЭСЮ) и саркомой Юинга кости имеются четкие различия. ЭСЮ чаще встречается у лиц старше 35 лет или младше 5 лет, средний возраст которых выше, чем у людей с саркомой Юинга кости.
Близкие расстройства
Симптомы следующих расстройств могут быть похожи на симптомы саркомы Юинга. Сравнения могут быть полезны для дифференциальной диагностики:
- Остеосаркома — опухоль, поражающая кости. Это самая распространенная форма рака костей. Примерно 60 процентов случаев происходят у детей и подростков в течение второго десятилетия жизни. Остеосаркомы поражают мужчин вдвое чаще, чем женщин. Чаще всего поражаются кости — длинные кости рук и ног. Симптомы могут варьироваться в зависимости от места и степени заболевания. Боль, отек, нежность и в конечном итоге образование комка может произойти в пострадавшем районе. Общие симптомы могут включать жар, потерю веса, анемию и недостаток энергии. Остеосаркомы могут ослабить окружающую кость, что приводит к переломам. Остеосаркомы могут распространяться (метастазировать) в другие области тела. Точная причина остеосаркомы неизвестна.
- Дополнительные опухоли также следует дифференцировать от саркомы Юинга, включая:
- хондросаркомы;
- остеохондромы;
- медуллобластомы;
- нейробластомы;
- рабдомиосаркомы;
- лимфомы костей.
- Остеомиелит — инфекция костей, обычно вызываемая бактериями. Остеомиелит может быть острым или хроническим. Расстройство обычно возникает в результате инфекции в одной части тела, которая транспортируется через кровоток к кости в отдаленном месте. Среди детей и подростков чаще всего поражаются длинные кости ног и рук. У взрослых остеомиелит чаще всего поражает позвонки позвоночника и/или бедро. Первоначально может быть несколько дней лихорадки и общее чувство плохого здоровья (недомогание). Остеомиелит может сопровождаться повышением температуры, глубокой локализованной болью в костях, ознобом, потливостью, отеком и болезненным или ограниченным движением близлежащих суставов. Кожа рядом с пораженной костью может быть красной (эритема), и может наблюдаться гной, разрушение окружающей ткани (некроз) и повреждение или деформация кости.
- Эозинофильная гранулема является подразделением редкого спектра расстройств, известных как гистиоцитоз из клеток Лангерганса (ГКЛ). ГКЛ характеризуется перепроизводством (пролиферацией) и накоплением определенного типа лейкоцитов (гистиоцитов) в различных тканях и органах организма. Они могут включать определенные отличительные гранулосодержащие клетки (клетки Лангерганса), участвующие в определенных иммунных реакциях, а также другие лейкоциты (например, моноциты, эозинофилы). У большинства людей с ГКЛ развиваются единичные или множественные поражения кости (эозинофильные гранулемы), вызванные ненормальным накоплением клеток Лангерганса и эозинофилов. В некоторых случаях эти поражения могут не сопровождаться какими-либо симптомами. Однако в большинстве случаев поражения связаны с болями в костях и отеком соседних тканей. Во многих случаях, также может произойти потеря кальция в костях (остеолиз). Чаще всего поражаются череп, позвоночник и длинные кости рук и ног. Также могут возникнуть вторичные осложнения, включая спонтанные переломы длинных костей или позвоночный коллапс и сдавление спинного мозга.
Диагностика
Диагноз опухоли семейства Юинга основывается на тщательной клинической оценке, выявлении характерных симптомов и физических данных, подробном анамнезе пациента и различных специализированных тестах. Такое тестирование включает в себя микроскопическую оценку опухолевых клеток и пораженной ткани (гистопатология) и молекулярный анализ в поисках транслокации EWS-FLI1.
— Клиническое тестирование и обследование.
Первоначально может быть проведена рентгенограмма, особенно если есть ощутимая масса. Рентген используется для получения изображений опухоли или пораженного участка. Более специализированные методы визуализации могут быть использованы для оценки размера, расположения и распространения опухоли (например, в мягкие ткани или костный мозг), чтобы определить, распространилась ли опухоль (метастазировала) в другие области тела (например, легкие и другие кости), и служить в качестве помощи для будущих хирургических процедур. Такие методы визуализации могут включать в себя компьютерную томографию (КТ), магнитно-резонансную томографию (МРТ) и сканирование костей. Биопсия костного мозга может показать, распространилась ли опухоль на костный мозг.
Диагноз саркомы Юинга может быть поставлен путем хирургического удаления (биопсии) и микроскопической оценки части пораженной ткани. Специализированный поверхностный белок, известный как CD99, обнаружен в большинстве опухолей семейства опухолей Юинга. Обнаружение присутствия этого белка может помочь в постановке диагноза саркома Юинга.
Стандартные методы лечения
Терапевтическое ведение пациентов с саркомой Юинга может потребовать скоординированных усилий команды медицинских специалистов, таких как врачи, которые специализируются на диагностике и лечении рака у детей (детские онкологи), взрослые онкологи, специалисты по применению радиации для лечения рак (радиационные онкологи), хирурги (ортопеды), онкологические медсестры и другие специалисты (в зависимости от места первичной опухоли).
Конкретные терапевтические процедуры и вмешательства могут варьироваться в зависимости от многочисленных факторов, таких как локализация первичной опухоли, степень первичной опухоли (стадия) и степень злокачественности; распространилась ли опухоль на лимфатические узлы или отдаленные участки; возраст человека и общее состояние здоровья; и/или другие элементы. Решения, касающиеся использования определенных вмешательств, должны приниматься врачами и другими членами команды здравоохранения при тщательной консультации с пациентом, исходя из особенностей его случая; тщательном обсуждение потенциальных выгод и рисков; предпочтения пациента; и других соответствующих факторов.
Лицам, страдающим саркомой Юинга, и членам их семей рекомендуется обращаться за консультацией после постановки диагноза и до начала лечения, так как диагноз может вызвать тревогу, стресс и крайние психологические расстройства. Пострадавшим от саркомы и их семьям рекомендуется психологическая поддержка и консультирование как на профессиональном уровне, так и в группах поддержки.
Лица с опухолью в семействе опухолей Юинга лечатся несколькими противоопухолевыми препаратами (химиотерапия) в сочетании с хирургическими процедурами и/или облучением. Хирургическое удаление злокачественной опухоли и пораженной ткани или облучения используется для лечения первичной опухоли. Химиотерапия убивает раковые клетки в первичном месте, а также скрытые раковые клетки, которые могли распространиться в другие области тела. Как правило, системная химиотерапия проводится в первую очередь, затем хирургическое вмешательство или облучение. Операция или лучевая терапия без адъювантной химиотерапии была намного менее эффективной, чем комбинированная терапия. Часто для лечения неоперабельных опухолей, а иногда и при метастазировании используется облучение.
Врачи используют несколько химиотерапевтических препаратов, поскольку разные препараты имеют разные способы действия при разрушении опухолевых клеток и/или предотвращении их размножения. Препараты химиотерапии, часто используемые для лечения пациентов с саркомой Юинга, включают доксорубицин, винкристин, циклофосфамид, дактиномицин, ифосфамид и этопозид.
Прогноз
По данным Американского онкологического общества, общая пятилетняя выживаемость при локализованной саркоме Юинга составляет 70%. Пациенты с метастатической болезнью имеют пятилетнюю выживаемость 15-30%.
Читайте также: