
Рефрактерная анемия дифференциальная диагностика

Людмила Смирнова,
заведующая кафедрой клинической гематологии и трансфузиологии БелМАПО,
доктор мед. наук,профессор.
Анемический синдром является самым распространенным гематологическим синдромом. Многие заболевания системы крови, а также других органов и систем (печени, почек, ЖКТ, иммунной системы, соединительной ткани), злокачественные опухоли различных локализаций могут начинаться с анемического синдрома. Снижение показателей гемоглобина менее 120 г/л у женщин фертильного возраста, менее 110 г/л у беременных, менее 130 г/л у мужчин выступает общепризнанным критерием анемического синдрома.
ИДЕАЛЬНОЙ КЛАССИФИКАЦИИ анемий не существует. Так, самую распространенную железодефицитную можно отнести к разным группам анемий: связанная с кровопотерей, нарушением кроветворения, опухолевым процессом и т. п. В связи с этим классификация по этиологическому принципу слишком громоздкая и неудобная для практического применения.
Андрей Воробьев и Лев Идельсон предложили рабочую классификацию анемий — патогенетическую, приемлемую для практического врача:
1) острые постгеморрагические;
2) железодефицитные;
3) связанные с нарушением синтеза или утилизации порфиринов (сидероахрестические);
4) связанные с нарушением синтеза ДНК и РНК (мегалобластные);
5) гемолитические;
6) связанные с нарушением пролиферации клеток костного мозга.
Каждая из этих групп анемий подразделяется на несколько подгрупп (или даже несколько десятков подгрупп, как, например, гемолитические).
На первом этапе распознавания анемии удобно использовать ИНДЕКСЫ КРАСНОЙ КРОВИ, предоставляемые гематологическим анализатором:
МСН — среднее содержание гемоглобина в одном эритроците. По этому критерию все анемии делятся на нормохромные (28–31 пикограмм (pg) в одном эритроците), гипохромные
( 31 pg). Нормальный показатель МСН характерен для острой постгеморрагической, гемолитической и апластической анемий;
МСV — средний объем эритроцита, нормальные границы 78–98 фемтолитров (fl). По данному показателю анемии делятся на нормоцитарные (78–98 fl), микроцитарные ( 98 fl);
RDW — широта распределения эритроцитов по объему, нормальные границы 11–14 %, выступает показателем анизоцитоза эритроцитов.
Цветовой показатель (ЦП) – устаревший индекс красной крови – показывает, является ли содержание гемоглобина в эритроцитах нормальным, повышенным или пониженным (норма 0,8–1,05). Данный показатель дает большую погрешность, его следует исключить из клинико-лабораторной практики.
Если выявляется гипохромная микроцитарная анемия (МСН 100 fl, MCH > 31 pg) можно предположить МЕГАЛОБЛАСТНУЮ АНЕМИЮ. В группу мегалобластных анемий (т. е. связанных с нарушением синтеза ДНК и РНК, сопровождающихся наличием в костном мозге патологических клеток красного ряда — мегалобластов) входят витамин В12-дефицитная (приобретенная и наследственная), фолиеводефицитная, а также ферментопатии (нарушение активности ферментов, участвующих в синтезе пуриновых или пиримидиновых оснований). Клинически для В12-дефицитной анемии характерны три синдрома: анемический, неврологический, поражение ЖКТ.
В периферической крови макроцитоз эритроцитов (MCV 100–160 fl, диаметр мегалоцита 12 мкм и более), отмечаются тельца Жолли, кольца Кебота, базофильная пунктация эритроцитов-макроцитов. Имеют место умеренная лейкопения (за счет нейтропении) и тромбоцитопения, что связано с неэффективным гранулоцитопоэзом и тромбоцитопоэзом. Отмечаются макроформы нейтрофилов, их гиперсегментация, снижение количества палочкоядерных, относительный лимфоцитоз. Уровень ретикулоцитов снижен ( 200 мкг/л и особенно > 500 мкг/л необходимо выполнить миелограмму после исключения мегалобластной анемии, лейкоза, целесообразна окраска мазка на берлинскую лазурь для подсчета сидеробластов, обязателен онкопоиск).
Оптимальные тесты для скрининга анемического синдрома — выявить уровень гемоглобина и СФ.
В практике терапевта анемический синдром встречается в определенных ситуациях, в большинстве случаев терапевт имеет дело с симптоматическими анемиями. Знание характера анемического синдрома и сочетающихся с ним заболеваний облегчает ориентировку на начальном этапе диагностики (см. схему).
Схема
Зависимость характера анемии от патологии
Следует иметь в виду, что при миелодиспластических синдромах (МДС) не существует патогномоничных признаков, и диагноз устанавливают с учетом анамнеза, данных осмотра, но прежде всего на основании комплекса лабораторных исследований. К обязательным методам обследования при установлении диагноза миелодиспластического синдрома (МДС) относятся:
• общий клинический анализ крови;
• морфологическое исследование мазков крови с подсчетом лейкоцитарной формулы и числа ретикулоцитов;
• морфоцитохимическое исследование аспирата костного мозга, включая реакцию Перлса, реакцию на ЩФ, миелопероксидазу, неспецифическую эстеразу, липиды;
• морфологическое исследования трепанобиопта-та костного мозга;
• цитогенетическое исследование клеток костного мозга и крови;
• при необходимости — иммунофенотипирование аспирата или материала трепанобиопсии костного мозга, проведение пробы Кумбса, определение уровня тромбоцитассоциированных антител, уровня эндогенного эритропоэтина, ЛДГ, исследование на ВИЧ-инфекцию и вирусные гепатиты, проведение сахарозной пробы и пробы Хема, определение уровня железа сыворотки и ферритина (которое необходимо для контроля терапии препаратами, влияющими на обмен железа).
Ввиду того, что ХММЛ ранее рассматривался в группе МДС и в связи с возможными дифференциально диагностическими трудностями, мы приводим дигностические критерии миелодиспластических/миелопролиферативных заболеваний за исключением ЮММЛ, поскольку он встречается в детском возрасте. Диагностическими критериями ХММЛ являются:
1) абсолютный моноцитоз в крови (более 1,0•10 9 /л);
2) отсутствие филадельфийской хромосомы или слитного гена BCR/ABL;
3) менее 20 % бластных клеток в крови или в костном мозге (бластные клетки включают миелобласты, монобласты и промоноциты, последние рассматриваются как эквивалент бластных клеток);
4) дисплазия клеток одного и более ростков кроветворения. При отсутствии дисплазии или ее минимальных проявлениях диагноз ХММЛ может быть установлен, если обнаружены:
а) клональные цито-генетические или молекулярно-биологические аномалии клеток костного мозга или
б) моноцитоз длительностью более 3 мес при условии исключения любых других причин моноцитоза, например опухоли или инфекции.
В связи с разным прогнозом в зависимости от числа бластных клеток ХММЛ разделен на два подварианта: ХММЛ-1 (с числом бластных клеток в крови менее 5 %, в костном мозге — менее 10%), ХММЛ-2 (с числом бластных клеток в крови от 5 до 19 %, в костном мозге — от 10 до 19 %). При наличии палочек Ауэра в бластных клетках и количестве бластов в крови и костном мозге менее 20 % устанавливается диагноз ХММЛ-2. Случаи ХММЛ с эо-зинофилией выше 1,5• 109/л классифицируют как ХММЛ-1 или ХММЛ-2 с эозинофилией.

Дифференциально-диагностические сложности могут возникать между миелодиспластическим синдромом (МДС) и АХМЛ. Диагноз АХМЛ устанавливают при сочетании следующих признаков:
• лейкоцитоз, обусловленный увеличением числа зрелых и незрелых нейтрофилов;
• выраженные признаки дисгранулопоэза;
• отсутствие филадельфийской хромосомы или слитного гена BCR/ABL;
• незрелые формы нейтрофилов (промиелоциты, миелоциты, метамиелоциты) составляют 10 % от общего числа лейкоцитов или более;
• отсутствие или незначительно выраженное увеличение числа базофилов, которые составляют менее 2 % от общего числа лейкоцитов;
• отсутствие или наличие незначительно выраженного абсолютного моноцитоза при числе моноцитов менее 2 % от общего числа лейкоцитов;
• гиперклеточный костный мозг с гиперплазией и дисплазией гранулоцитарного ростка с диспла-зией эритроидного и мегакариоцитарного ростков или без нее;
• менее 20 % бластных клеток в крови и костном мозге.
Частота обнаружения цитогенетических аномалий при АХМЛ может достигать 80 %, однако специфичные аберрации отсутствуют. Прогноз неблагоприятный: медиана выживаемости не превышает 20 мес, а до трансформации заболевания в острый лейкоз доживают от 25 до 40 % больных.
К признакам МДС/МПЗ-Н относят:
• случаи, сопровождающиеся клиническими, лабораторными и морфологическими признаками какого-либо варианта МДС (рефрактерная анемия, рефрактерная анемия с кольцевыми сидеробластами, рефрактерная цитопения с мультилинейной дисплазией, рефрактерная анемия с избытком бластов) при числе бластных клеток в крови и костном мозге менее 20 % и
• выраженные признаки миелопролиферации, например тромбоцитоз 600•10 9 /л и выше в сочетании с пролиферацией мегакариоцитов или лейкоцитоз 13,0•10 9 /л и выше с выраженной спленомегалией или без нее и
• отсутствие в анамнезе хронических миелопролиферативных заболеваний или МДС, отсутствие недавно проведенного лечения цитотоксическими или ростовыми факторами, что может объяснить признаки миелодисплазии или миелопролиферации, а также отсутствие филадельфийской хромосомы или слитного гена BCR/ABL, t(3;3)(q21;q26) или inv(3)(q21q26) или
• наличие у больного смешанных миелопролиферативных и миелодиспластических признаков и невозможность отнесения заболевания ни к одной другой категории МДС, хронического миелопролиферативного заболевания или хронического миелодиспластического/миелопролиферативного заболевания.
Трудности диагностики миелодиспластических синдромов обусловлены тем, что морфологические изменения не являются специфическими для МДС и встречаются при других патологических состояниях неопухолевой природы. В частности, они характерны для В12 и фолиеводефицитной анемий, возможны при интоксикации тяжелыми металлами, алкогольном поражении печени, различных воспалительных процессах, пароксизмальной ночной гемоглобинурии (ПНГ), наследственной форме болезни Пельгера, СПИДе, а также как следствие проводимой цитостатической терапии.
Дифференциальный диагноз гипопластического варианта миелодиспластического синдрома (МДС) проводится с апластической анемией (АА). В первом случае в материале трепанобиопсии костного мозга наблюдается пролиферация трех ростков кроветворения и может выявляться АЛМП. При гипопластическом миелодиспластического синдроме значительно реже происходит увеличение числа тучных клеток. При АА в большинстве случаев определяется только одноростковая (эритроидная) пролиферация без признаков дисплазии гемопоэза. Относительная гиперплазия мегакариоцитов характерна для миелодиспластического синдрома, в то время как при АА наблюдается их выраженная гипоплазия или аплазия.
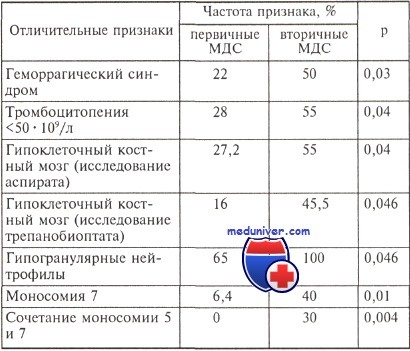
В исследовании I. Lorand-Metze и соавт. в трепанобиоптатах при апластической анемии мегакариоциты, которые авторы выявляли иммуногистохимически, отсутствовали у 12 из 20 больных, в то время как при гипопластическом миелодиспластическом синдроме — у 1 из 21.
При апластической анемии также было значительно ниже среднее число мегакариоцитов, а основная часть гемопоэтических клеток была представлена нормобластами и лимфоцитами. При миелодиспластическом синдроме определялась большая неравномерность участков костного мозга, содержащих кроветворные клетки. При иммуногистохимическом исследовании трепанобиоптатов больных с гипопластическим вариантом миелодиспластического синдрома выявлено достоверно большее число CD34+-клеток по сравнению с АА (0,94 и 0,04 % соответственно), а также большее число PCNA-(proliferating cell nuclear antigen — ядерный антиген пролиферирующих клеток) позитивных клеток, чем при АА (43,59 и 14,80 % соответственно).
Важным методом исследования, позволяющим диагностировать МДС, является цитогенетическое исследование, которое позволяет обнаружить характерные хромосомные аномалии. Однако имеются работы, указывающие на возможность изменений кариотипа и при АА, например, +6 или t(l;20). Тем не менее в большинстве случаев при АА хромосомные изменения выявляются при длительном течении болезни, либо после иммуносупрессивного лечения (например, моносомия хромосомы 7), либо после трансформации АА в миелодиспластический синдром (МДС) или ОЛ. При первичном обследовании больных АА частота аномалий кариотипа не превышает 4 %.
Дифференциальный диагноз при фиброзном варианте миелодиспластического синдрома (МДС) проводится с хроническими миелопро-лиферативными заболеваниями, наиболее часто — с хроническим идиопатическим миелофиброзом (ХИМФ). Последний за редким исключением протекает со спленомегалией, лейкоцитозом со сдвигом формулы крови влево до миелоцитов и тромбоцитозом. В крови и костном мозге отсутствуют многочисленные проявления дисплазии гемопоэтических клеток. При исследовании мазка крови зачастую выявляют нормоцитоз, пойкилоцитоз с каплевидной формой эритроцитов, ядра и фрагменты мегакариоцитов. ХИМФ характеризуется более выраженным фиброзом костного мозга, в том числе коллагеновым.
Цитогенетическое исследование иногда позволяет выявить менее характерные для миелодиспластического синдрома изменения: трисомия хромосомы 21, утрата хромосомы 13 или ее длинного плеча. Дополнительным признаком, указывающим на ХИМФ, является повышенный уровень ЩФ гранулоцитов (однако и этот признак нередко обнаруживается при миелодиспластическом синдроме).
Необходимость дифференциального диагноза с ПНГ обусловлена общей для обоих заболеваний панцитопенией при нормо- или гиперклеточном костном мозге. Отличительным признаком ПНГ является ретикулоцитоз крови, положительные пробы Хема и сахарозная, обнаружение гемосидерина в моче и свободного гемоглобина в сыворотке крови.
Иногда возникают трудности в дифференциальном диагнозе ХММЛ и ХМЛ. Как известно, при ХМЛ обнаруживается дисплазия мегакариоцитарного ростка, иногда имеются черты дисплазии и в гранулоцитарном ростке. В связи с наличием лейкоцитоза может быть увеличено абсолютное число моноцитов, в крайне редких случаях в хронической фазе и значительно чаще в фазе акселерации выявляется тромбоцитопения. При исследовании трепанобиоптата в хронической фазе ХМЛ у 40 % больных может обнаруживаться ретикулиновый фиброз, а в фазе акселерации — и коллагеновый фиброз. Однако в большинстве случаев нарастающий лейкоцитоз с характерными изменениями лейкоцитарной формулы и обнаружение t(9;22) и/или слитного гена BCR/ABL при цитогенетическом исследовании позволяют установить правильный диагноз.
Теперь перейдем к частной патологии анемического синдрома. В этом же подразделе разберем дифференциальную диагностику различных анемических состояний, основное место в которой занимает гематологический фактор (т.е. лабораторные показатели).
Острая постгеморрагическая анемия – наблюдается при потере большого количества крови за короткий промежуток времени. Минимальная одноразовая кровопотеря, представляющая опасность для здоровья взрослого человека – 500 мл.
Этиология. Причинами острой постгеморрагической анемии могут быть внешние травмы или кровотечения из внутренних органов: после массивных кровотечений из расширенных вен пищевода, сосудов желудка или 12-перстной кишки при язвенной болезни, из язвы тонкой кишки при брюшном тифе, при разрыве маточной трубы в случае внематочной беременности, разъедании ветви легочной артерии при туберкулезе легких, разрыве аневризмы аорты или ранении ее стенки и отходящих от аорты крупных ветвей. Чем крупнее калибр пораженных сосудов и чем ближе он расположен к сердцу, тем опаснее для жизни кровотечение. При разрыве дуги аорты достаточно потерять 1 л крови, чтобы наступила смерть. Смерть наступает в таких случаях прежде, чем происходит обескровливание органов из-за резкого падения артериального давления и дефицита наполнения полостей сердца. При кровотечении из сосудов мелкого калибра смерть наступает при потере более половины общего количества крови.
Патогенез постгеморрагической анемии связан с острой сосудистой недостаточностью за счет резкого уменьшения ОЦК и снижением кислородно-транспортной функции крови (гипоксии) вследствие потери эритроцитов, когда эта потеря уже не может быть компенсирована за счет учащения сердечных сокращений (компенсаторной тахикардии).
Клиническая картина. У больного прежде всего появляются симптомы коллапса: резкая слабость, головокружение, бледность, сухость во рту, холодный пот, рвота, падает артериальное и венозное давление, уменьшается сердечный выброс крови, резко учащается пульс, наполнение пульса становится слабым. Определенное значение имеет источник кровопотери: к примеру, кровотечение в желудочно-кишечный тракт может сопровождаться повышением температуры тела, картиной интоксикации и т.п.
Как уже было сказано, клиническая картина и тяжесть состояния пациента определяются не только количеством потерянной крови, но и скоростью кровопотери, калибром пораженного сосуда.
Картина крови. В периферической крови сразу после кровотечения цифровые показатели эритроцитов и гемоглобина мало отличаются от исходных. Анемия выявляется через 1–2 дня. К этому времени объем крови восполняется за счет поступления в сосуды тканевой жидкости. В периферической крови увеличивается количество ретикулоцитов и юных эритроцитов (нормобластов). Из-за дефицита железа уровень гемоглобина снижен.
Хроническая постгеморрагическая анемия – чаще всего это вариант ЖДА (разбор впереди).
Картина крови:
- равномерное снижение количества эритроцитов и гемоглобина
- умеренный нейтрофильный лейкоцитоз и ретикулоцитоз
- увеличение числа тромбоцитов
Железодефицитные анемии (ЖДА).Обусловлены нехваткой железа в организме, что приводит к множественным трофическим нарушениям (ломкость ногтей, сухость кожи, выпадение волос, снижение мышечной силы), а затем к недостаточному образованию гемоглобина в клетках эритроидного ряда, что проявляется гипохромной анемией.
Этиология и патогенез. Основная часть поступающего в организм железа расходуется на образование гемоглобина. Депонирование осуществляется за счет связывания со специфическими белками в форме ферритина и гемосидерина. Трансферрин является транспортным белком: благодаря ему депонированное или всасывающееся в ЖКТ железо доставляется к клеткам, синтезирующим гемоглобин. У здоровых мужчин в запасе находится примерно 1000 мг (1г) железа. У женщин количество депонированного железа может варьировать от 0 до 500 мг. Примерно у 1/3 клинически здоровых женщин запасы железа в депо вовсе отсутствуют. Всасывание железа осуществляется во всех отделах ЖКТ, однако наиболее эффективно – в двенадцатиперстной кишке. У здоровых людей из пищи всасывается примерно 10% железа. Чаще всего железодефицитные анемии наблюдаются у женщин в связи с тем, что потеря железа у них примерно в 2 раза выше, чем у мужчин. Основная причина избыточной потери железа – менструальные кровотечения (меноррагии) и рождение детей.
Причиной дефицита железа может стать неполноценная диета (голод, невозможность или отказ от употребления мяса). Особую группу составляют больные с тяжелым поражением тонкого кишечника, при котором нарушается всасывание железа. Дефицит железа нередко развивается после резекции тонкой кишки вследствие кровотечений из ЖКТ (геморрой, язва желудка или двенадцатиперстной кишки, грыжа пищеводного отверстия диафрагмы, ангиоматоз, дивертикулез или полипоз толстого кишечника, неспецифический язвенный колит, болезнь Крона).
Клиническая картина. Характерны вялость, повышенная утомляемость, головная боль, сухость кожи и другие трофические нарушения, причем эти симптомы появляются у больного еще до развития выраженной анемии.
Характерным является сидеропенический синдром – снижение аппетита, затруднение при глотании твердой пищи, извращения вкуса в виде желания есть мел, глину, уголь и т.п., появляется пристрастие к необычным запахам (керосин, гуталин, ацетон).
Отмечается ломкость, искривление, поперечная исчерченность ногтей. Определяются бледность кожи, слизистых оболочек, сердцебиение, часто выслушивается “невинный” систолический шум, беспокоит одышка при физической нагрузке. Примерно у 10% больных пальпируется селезенка.
Важное значение в клинической картине имеет характер заболевания, определившего дефицит железа (язвенная болезнь желудка и 12-перстной кишки, геморрой, миома матки и т.п.) В связи с этим необходимо отметить, что многие хронические внутренние кровотечения могут оставаться скрытыми в течение многих лет, несмотря на многократные рентгенологические, гастро-, дуодено- и колоноскопические исследования.
Дифференциальный диагноз проводят с анемией, наблюдающейся при хронических воспалительных процессах и онкологических заболеваниях.
Картина крови:
- гипохромия (ЦП ниже 1,0)
- микроцитоз (см. классификацию)
- снижение содержания сывороточного железа (ниже 12 ммоль/л)
- может быть ускорение СОЭ
Анемии, связанные с нарушением синтеза или утилизацией порфиринов (сидероахрестические).Подобные анемии бывают наследственными и приобретенными.
Наследственными формами сидероахрестической анемиичаще болеют мужчины. Заболевание наследуется по рецессивному типу и передается с X-хромосомой.
Патогенез: в большинстве случаев обнаруживается снижение содержания протопорфирина эритроцитов, что обусловливает невозможность связывания железа. Вследствие этого происходит накопление железа в организме при резко нарушенном образовании гемоглобина.
Клиническая картина. Заболевание начинается с детства или с юности. Жалобы могут отсутствовать, иногда больные жалуются на небольшую слабость, повышенную утомляемость. Печень увеличена, иногда увеличена и селезенка. При выраженных отложениях железа в тех или иных органах развивается соответствующая клиника. Если железо поступает преимущественно в печень, развивается цирроз (гемосидероз печени). При отложении в поджелудочной железе может развиться сахарный диабет. Накопление железа в сердечной мышце приводит к недостаточности кровообращения, в яичках — к развитию евнухоидизма. Иногда кожа приобретает серый оттенок. Характерным является отсутствие клинических признаков ЖДА (нет астенизации, сухости кожи, выпадения волос, деформации ногтей и т.п.) при наличии выраженной гипохромной анемии.
Картина крови: цветовой показатель — 0,4–0,6 (снижен). Эритроциты в мазке гипохромные, повышен уровень сывороточного железа. Но окончательный диагноз может быть выставлен при исследовании уровня порфиринов в эритроцитах.
Приобретенные формы сидероахрестической анемии связаны с попаданием большого количества свинца в организм. Свинец блокирует сульфгидрильные группы ферментов, участвующие в синтезе гемоглобина.
Для свинцовой интоксикации типично сочетание признаков: мишеневидные эритроциты в мазке крови, высокое содержание железа в сыворотке крови, повышенный гемолиз эритроцитов, боли в животе, симптомы полиневрита.
Характерным признаком является развитие гипохромной анемии при высоком уровне сывороточного железа, а также наличие клинических и анамнестических признаков свинцовой интоксикации.
Витамин В12 – дефицитная анемия -по другому – пернициозная анемия, болезнь Аддисона-Бирмера; по картине крови – мегалобластическая (мегалобластная). Заболевание характеризуется появлением в костном мозге мегалобластов, внутрикостномозговым разрушением эритроцитов, гиперхромной макроцитарной анемией, тромбоцитопенией и нейтропенией, атрофическими изменениями слизистой оболочки ЖКТ и изменениями нервной системы в виде фуникулярного миелоза.
Этиология и патогенез. Идиопатическая форма В12-дефицитной анемии (пернициозная анемия) развивается в результате недостаточного поступления в организм экзогенного цианокобаламина преимущественно у лиц пожилого возраста. Патогенез дефицита витамина В12 чаще связан с нарушением выработки гликопротеина (внутреннего фактора), соединяющегося с пищевым витамином В12 (внешний фактор Кастла) и обеспечивающего его всасывание. Нередко первые признаки заболевания появляются после перенесенного энтерита, гепатита. В первом случае это обусловлено нарушением всасывания витамина в тонкой кишке, во втором – расхождением его запасов в печени, являющейся основным депо витамина В12. Развитие В12-дефицитной анемии после тотальной гастрэктомии (когда полностью ликвидируется секреция внутреннего фактора) происходит через 5-8 лет и более после операции. В течение этого срока больные живут запасами витамина в печени при минимальном пополнении его за счет очень незначительного всасывания в тонкой кишке, не соединенного с внутренним фактором Кастла.
Более редкими причинами нарушенного выделения внутреннего фактора может быть хроническая алкогольная интоксикация, когда она сопровождается токсическим поражением слизистой оболочки желудка или при инвазии широким лентецом, когда паразит поглощает большое количество витамина В 12.
Картина крови:
- гиперхромия эритроцитов (ЦП выше 1,0)
- пониженное количество эритроцитов
- макроцитоз (мегалоцитоз или мегалобластоз – это практически синонимы)
- тромбоцитопения и лейкопения
- появление мелких шизоцитов
Но решающее значение в диагностике имеет исследование костного мозга, которое обнаруживает резкое увеличение в нем мегалобластов.
Фолиеводефицитная анемия – та же мегалобластная анемия, по сути почти В12- дефицитная. Существенная разница заключается в том, что болезнью Аддисона – Бирмера страдают взрослые дяди (за 40 лет), а типичным для развития фолиеводефицитной анемии является младший детский возраст, так что эта нозология – прерогатива педиатров, но мы с нею позднее познакомимся.
Гемолитические анемии -включают в себя целую группу нозологических единиц, объединяющим признаком которых является преобладание процессов кроворазрушения над процессами кровеобразования. Кроворазрушение может происходить преимущественно внутри сосудов или вне сосудов. Причины внутрисосудистого гемолиза: гемолитические яды, тяжелые ожоги, малярия, сепсис, переливание несовместимой крови, иммунопатологические процессы, вирусные инфекции, хронический лимфолейкоз, системная красная волчанка, пароксизмальная холодовая гемоглобинурия. Причины внесосудистого гемолиза носят семейный наследственный характер.
Гемолиз эритроцитов имеет место прежде всего в селезенке. Он может быть связан с дефицитом ферментов (глюкозо-6-фосфат-дегидрогеназы, пируваткиназы); с нарушением синтеза гемоглобина.В нашей стране наиболее распространенной гемолитической анемией является наследственный сфероцитоз.
Этиология: заболевание передается генетическим путем, но с полом не связано.
Патогенез. Дефект структуры мембран эритроцитов приводит к потере ими нормальной формы двояковогнутого диска и приобретению формы шара (сферуляция). Подобные клетки быстро разрушаются в селезенке. Вследствие ранней гибели эритроцитов уменьшается их количество. Сферуляция эритроцитов происходит только в периферической крови, а не в костном мозге.
Клиническая картина. У больных развивается желтуха, увеличивается селезенка, большая или меньшая степень анемии, склонность к образованию камней в желчном пузыре. Если болезнь развивается в раннем детстве, имеет место деформация скелета, особенно черепа. Череп приобретает башенную квадратную форму, отмечается микроофтальмия, изменяется расположение зубов, выявляется высокое небо. У ряда больных имеет место укорочение мизинца. Гемолитические кризы чаще всего провоцируются инфекцией.
Картина крови. В периферической крови увеличено число ретикулоцитов до 10 %, иногда до 50–60 %. В мазке крови сфероциты выглядят в виде маленьких клеток с интенсивной окраской, без обычной для нормальных эритроцитов бледности в центре. Относительное число сфероцитов — в среднем 20– 30 %, но нередко они составляют большинство красных клеток.
Дифференциальный диагноз. Следует проводить с аутоимунной гемолитической анемией. В этом случае помогает анамнез, наличие изменений скелета (башенный череп, короткие мизинцы), выявление аутоантител.
Апластические анемии.Заболевание, характеризующееся панцитопенией периферической крови и снижением содержания клеток костного мозга.
Этиология и патогенез. Апластическая анемия – относительно редкое заболевание. Встречается с частотой примерно 0,5 на 100 000 населения. Число случаев болезни возрастает по мере увеличения возраста от 1 года до 20 лет. Существенных различий в заболеваемости от 20 до 60 лет не выявлено. После 60 лет число случаев болезни возрастает. В некоторых семьях отмечается генетическая предрасположенность к болезни. Есть сообщения о развитии аплазии после приема некоторых лекарственных препаратов. Примерно у 50% больных установить этиологию заболевания практически не удается (идиопатическая апластическая анемия).
Клиническая картина связана в основном с цитопеническим синдромом и зависит от его глубины. Чаще всего наблюдается геморрагический синдром, в особенности при снижении уровня тромбоцитов (петехиальные высыпания на коже, синячки, кровотечения из десен, носа, почек, матки; возможны тяжелые кровотечения из желудочно-кишечного тракта). Появление геморрагии на слизистой оболочке полости рта, кровоизлияния в конъюктиву являются грозными симптомами, свидетельствующими о весьма возможном кровоизлиянии в головной мозг. Наряду с геморрагическим синдромом в клинической картине превалируют инфекционные осложнения (бронхит, пневмония, сепсис, парапроктит). Возможны тяжелые грибковые поражения слизистых оболочек, грибковый сепсис. Наряду с геморрагическими и инфекционными осложнениями часто выявляются симптомы, связанные с глубокой анемией (слабость, одышка при физической нагрузке, стенокардия).
При проведении дифференциальной диагностики необходимо учитывать, что панцитопения может явиться первым признаком острого лейкоза, метастазов рака в костный мозг, костномозговой формы хронического лимфолейкоза, волосато-клеточного лейкоза, туберкулеза, гепатита вирусной этиологии. Диагноз “апластическая анемия” в принципе является диагнозом исключения и устанавливается лишь в том случае, если перечисленные выше причины развития аплазии отвергнуты.
Картина крови: - панцитопения (снижение эритрцитов, лейкоцитов, тромбоцитов). Для уточнения диагноза необходима стернальная пункция и исследование пунктата.
Читайте также: